Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Nutrition & Dietetics
Fanconi anemia (FA) is a rare disorder with the clinical characteristics of (i) specific malformations at birth, (ii) progressive bone marrow failure already during early childhood and (iii) dramatically increased risk of developing cancer in early age, such as acute myeloid leukemia and squamous cell carcinoma. Patients with FA show DNA fragility due to a defect in the DNA repair machinery based on predominately recessive mutations in 23 genes.
- Fanconi anemia
- epigenetics
- cancer
- immunology
1. Clinical Features of FA
FA was described first in 1927 by the Swiss pediatrician Guido Fanconi [1]. Clinically the disease is often summarized with the triad of congenital malformations, a progressive bone marrow failure and a dramatically increased risk of developing cancer [2]. This rare inherited multisystem disorder is mostly associated with a broad clinical spectrum consisting of typical malformations but can show in some cases also a completely normal phenotype [3][4]. FA-typical malformations present mainly in the skeletal system, especially at the radius/thumb [5]. Moreover, FA frequently affects the whole body, such as low body weight and reduced height, but also malformations in the skin (café au lait spots) [6] or in inner organs, such as heart, kidney and intestine, are common features of the disease [7]. In addition, approximately 80% of all FA patients show signs of an ineffective hematopoietic system and develop bone marrow failure, myelodysplastic syndrome or acute myeloid leukemia [2][8]. Declining blood counts affecting all blood lineages are often the first sign of these hematological features and commonly present already in the first decade of life [9]. Like in other syndromes associated with bone marrow failure, the myeloid system is more severely affected than the lymphoid system, which provides the patients with a high risk of acute infections.
In the past, progressive bone marrow failure was the main cause of death of FA patients [10]. Hematopoietic stem cell transplant is the only curative treatment for the hematological complications. Improved outcome of these transplants are the main reason why today’s FA patients have a higher life expectancy [11]. This is mainly due to higher donor availability, individual treatment protocols [12] and more advanced therapies against graft-versus-host disease and viral infections [13][14]. Moreover, better acute myeloid leukemia and myelodysplastic syndrome surveillance, such as frequent checks for chromosomal changes [15], drastically improved the identification of FA individuals at risk. At present, a treatment with synthetic testosterone analogs at supra-pharmacological doses accomplishes stabilization and increase of declining blood counts [16][17][18][19] but cannot prevent myelodysplastic syndrome or acute myeloid leukemia. However, mechanistically this treatment of the hematopoietic system is not well understood [20] and side effects can be therapy limiting [21].
Individuals with FA carry an enormous risk of developing squamous cell carcinoma, especially of the oral mucosa but also in the pharynx, larynx, esophagus, anus and vulva. Compared with the average population these cancers arise at much earlier age and there is a tendency for frequent syn- and meta-chronic squamous cell carcinomas [22]. Due to the genetic defect underlying the disease, treatment options are mostly limited to surgical removal of the cancer [23][24]. Thus, at present squamous cell carcinomas are the most life-threatening complications for adult FA patients. Despite the clinical significance of squamous cell carcinomas, there is still a lack of knowledge as to why FA patients have such an elevated risk of this type of cancer, which is rather uncommon in the general population. Premature aging, DNA fragility, endogenous and exogenous exposure to aldehydes, infections with the human papillomavirus and other chronic infections or inflammations have been discussed in this context but a clear mechanistic explanation is still lacking [25][26][27][28][29][30][31][32].
In addition to these life-threatening and limiting complications, an FA individual is facing numerous other clinical dysfunctions. FA patients have a wide range of metabolic and endocrine impairments [33][34] affecting lipid metabolism [35], glucose and/or insulin homeostasis [36], the thyroid axis [37] and most important fertility [38]. These non-cancerous aspects of FA often dramatically reduce the quality of life of the individuals.
2. Genetic and Molecular Features of FA
Until now, 23 genes have been identified that associate with FA, in the majority of which the mode of inheritance is autosomal recessive [39]. Patients with the complementation group R (FANCR) carry a heterozygous mutation in the RAD51 recombinase (RAD51) gene [39][40], whereas the FANCB gene is located on the X chromosome [41]. The main cellular function of FA genes is maintaining genomic integrity during DNA replication via intra-strand cross-linking repair and controlling the replication fork [42]. FA proteins are linked to homologous recombination conducting DNA repair; in the canonical pathway the so-called upstream FA core complex proteins activate the FANCI-FANCD2 complex via mono-ubiquitination [43], which promotes recruitment of DNA repair effectors to chromatin lesions, in order to resolve DNA damage and mitosis. Some of these downstream FA genes are known as tumor suppressor genes in other monoallelic inherited cancers like breast and ovarian cancer (FANCD1 = BRCA2, FANCS = BRCA1, FANCN = PABLB2, FANCJ = BRIP1, FANCO = RAD51C). Impairment in the FA pathway leads to increased spontaneous and inducible chromosomal fragility [44] and cell cycle arrest [45], which are both hallmarks of the cellular phenotype of FA.
In recent years, the molecular understanding of the role of FA proteins has rapidly grown in addition to functions in genomic maintenance and homeostasis mainly during replication. For example, FA proteins are linked to aldehyde detoxification [46][47] and altered selective autophagy, a key step in immunity, leading to increased mitochondrial reactive oxygen species-dependent inflammasome activation and mitophagy [48]. Moreover, altered mitochondrial functions [49][50][51] and increased oxidative stress [52][53] are linked to FA. This also implies direct interaction of FA proteins with altered insulin secretion [36] and lipid metabolism [54]. Furthermore, non-canonical functions of FA proteins in the control of cytokines, such as tumor necrosis factor [55] and transforming growth factor beta [56] have been described.
Taken together, the understanding of FA as a pure DNA damage repair disease shifts towards a more holistic view, shedding light on energy metabolism. Thus, the cellular and the clinical phenotype of FA can also be subsumed as a premature aging syndrome [30][57].
3. FA and Cancer
Based on its clinical and cellular phenotype FA can also serve as a cellular model for the study of general molecular functions and physiological aspects like aging as well as other non-communicable diseases occurring in the general population. In that respect, the study of FA had a considerable impact on the molecular understanding of breast/ovarian cancer [58]. Moreover, FA genes are also frequently mutated or dysregulated in sporadic cancers [59] as well as in childhood cancers [60]. Nevertheless, the enormous cancer risk of FA patients still needs to be elucidated mechanistically. Herein, the disturbances of the different FA genes represent the key intrinsic factors of the fragile system of FA individuals (Figure 1).
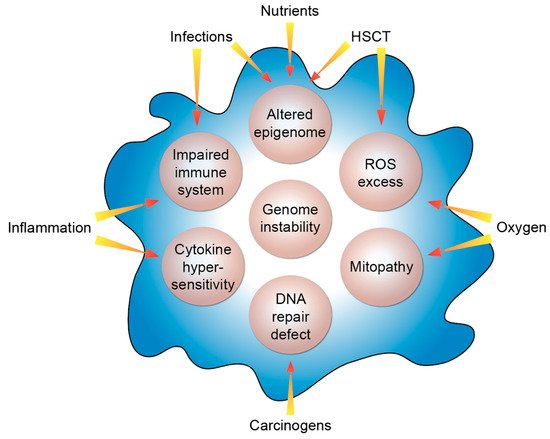
Figure 1. Molecular features of FA. The listed intrinsic processes (beige circles) are modulated by the indicated extrinsic factors. HSCT, hematopoietic stem cell transplant; ROS, reactive oxygen species.
The different clinical presentations of FA patients point out that a number of extrinsic modifiers have a significant modulatory impact on the course of the disease. Like in tumorigenesis, the disease modifiers do not need to be disease causing by themselves. Typical examples of such extrinsic factors can be oxygen, inflammation or infections like by human papillomavirus or Candida albicans. Additionally, carcinogens at low concentrations can tip the scale. Another example is hematopoietic stem cell transplant: FA patients exhibit an up to 700-fold increased risk for the development of squamous cell carcinomas compared to the general population [8] but in FA individuals that had received a hematopoietic stem cell transplant, this risk is even more elevated and squamous cell carcinomas occur in them at a younger age [61][62]. Risk analyses have identified graft-versus-host disease, i.e., a dysregulation of the transplanted immune system of the donor, as the underlying factor [63]. In balance of these negative extrinsic factors, nutrients, such as vitamin D, can act as positive extrinsic modifiers mainly via affecting the epigenome. Thus, intrinsic and extrinsic factors together determine the clinical individual cause of the disease.
Despite its rareness, the hematopoietic clonal disease and expansion in FA are intensely studied. Somatic amplifications at chromosomes 1q, 3q (including the gene MECOM (MDS1 and EVI1 complex locus)), and deletions of chromosome 7 during the aplastic phase of the disease display the origin of the respective clones [15][64][65][66]. In that respect, MECOM plays a crucial role as it encodes for a transcriptional regulator with an essential role in hematopoiesis and mediating epigenetic modifications by interacting with DNA, proteins and protein complexes [67]. Thus, the overexpression of MECOM provides the cell with growth advantages and disturbs the epigenetic landscape. Moreover, at the stage of myelodysplasia, de-regulations of the RUNX1 gene are frequently found [65]. Thus, the clonal expansion of such altered hematopoietic cells ultimately leads to myelodysplastic syndrome and acute myeloid leukemia [64]. Furthermore, in FA the changes on chromosomes 1, 3 and 7 are associated with a negative outcome after hematopoietic stem cell transplant [15]. Even though the association between these specific chromosomal changes and disease progression towards acute myeloid leukemia is well characterized, it is still not elucidated why and how those initial changes arise.
Naturally, studying negative disease modifiers is much easier than identifying and attributing the significance of preventive modifiers, such as vitamin D. Therefore, there is still a lack of knowledge in determining specific preventive factors besides a general healthy lifestyle, e.g., physical activity, healthy diet and the avoidance of smoking [68]. In summary, the occurrence of inherited mutated FA genes primarily indicates the fragility of the system “health”, while intrinsic and extrinsic factors are the real modifiers of the disease. As FA gene mutations cannot be changed in the whole body, the modulation of disease modifiers bears the potential of therapy and even disease prevention.
4. The Impact of Epigenetics in FA
The protein-DNA complex of histones and genomic DNA is referred to as chromatin [69]. The key function of chromatin is to keep most of the genome inaccessible to transcription factors and RNA polymerases, i.e., in a cell- and tissue-specific fashion chromatin functions as a gatekeeper for undesired gene activation. Differentiation processes are controlled by epigenetic programing, i.e., a change of the so-called epigenetic landscape composed of transcription factor binding, histone modifications and chromatin accessibility [70]. Thus, through epigenetics terminally differentiated cells have a permanent memory about their identity [71].
Next-generation sequencing techniques, which had been developed after the sequencing of the human genome, such as chromatin immunoprecipitation sequencing (ChIP-seq) and formaldehyde-assisted isolation of regulatory elements sequencing (FAIRE-seq), allow the genome-wide assessment of the transcription factor binding, histone modifications and chromatin accessibility [72]. These approaches have been systematically applied by large research consortia, such as ENCODE (www.encodeproject.org) and Roadmap Epigenomics (www.roadmapepigenomics.org), for the epigenome-wide characterization of more than one hundred human cell lines [73] and a comparable number of primary human tissues and cell types [74], respectively. It should be kept in mind that every single cell of an individual carries the same genome, but that there are hundreds to thousands different epigenomes, in which the tissues and cell types differ significantly.
The genomic region of the vitamin D target gene FANCE [75] serves as an illustrative example of vitamin D-triggered epigenetic changes in the context of FA (Figure 3). The FANCE gene encodes for a critical protein of the FA core complex mediating FANCD2/FANCI mono-ubiquitination, which is the essential activation step of the FA/breast cancer DNA-repair pathway [76]. In the monocytic cell line THP-1, which was derived from a 1-year old male patient with acute myeloid leukemia [77], ChIP-seq indicated a VDR binding site 9 kb downstream of the transcription start site of the FANCE gene. Within this enhancer region 1,25(OH)2D3 not only significantly increased the binding of VDR but also of its pioneer factor CEBPA. In parallel, at this genomic region the amount of accessible chromatin as well as the histone marker of active chromatin, H3K27ac, raised after treatment of the cells with the VDR ligand. Looping of this enhancer to the transcription start site of the FANCE gene results in 1,25(OH)2D3-triggered changes of accessible chromatin, H3K27ac markers and markers of active transcription start sites, H3K4me3. Taken together, vitamin D changes specifically on the level of VDR and CEBPA binding, chromatin markers and accessible chromatin of the epigenome at the region of the FANCE gene.
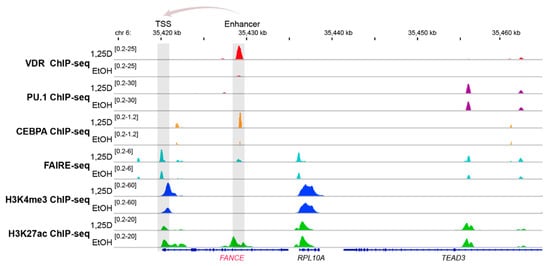
Figure 3. Vitamin D-triggered epigenomic profile in the region of the FANCE gene. The IGV browser [78] was used to display the epigenomic profiles at enhancer and transcription start site (TSS) regions of the vitamin D target gene FANCE. THP-1 cells had been treated for 24 h with 1,25(OH)2D3 (1,25D) or vehicle (EtOH) and in three biological repeats, ChIP-seq experiments had been performed with antibodies against VDR [79], the pioneer factors PU.1 [80] and CEBPA [81], the histone marker for active transcription start site (TSS) regions, H3K4me3 [82] and the marker for active chromatin, H3K27ac [82], such as at enhancers, as well as FAIRE-seq [75] for accessible chromatin. The gene structures are shown in blue and vitamin D target gene FANCE is indicated in red. The genes RPL10A and TEAD3 serve as non-regulated references.
In general, epigenetics associates with lifestyle and environmental conditions of healthy as well as of diseased individuals, such as FA patients [83]. The dynamic profile of the epigenome provides the advantage that some events of epigenetic programing are reversible. This implies that lifestyle changes can improve health and prevent or milden disease, such as complications of FA. Thus, as long as no irreversible tissue damage has happened, it is in the hands of the individual to reverse a disease condition. Accordingly, there is a high level of individual responsibility for staying healthy and epigenetics provides a molecular explanation for this life philosophy [71].
This entry is adapted from the peer-reviewed paper 10.3390/nu12051355
This entry is offline, you can click here to edit this entry!