Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
Vitamin D, conventionally considered a nutrient, is a potent hormone regulating many physiological functions. Vitamin D exists as a prohormone that needs to be transformed into biologically active products that bind to their cognate nuclear receptors to regulate diverse physiological processes.
- Vitamin D
- metabolic pathways
1. Canonical Vitamin D Metabolic Pathway
There are two major isoforms of vitamin D, vitamin D2 (ergocalciferol) and vitamin D3 (cholecalciferol) [29,30]. Both vitamin D2/3 need exposure to sunlight’s UVB radiation to be synthesized from ergosterol and 7-dehydrocholesterol, respectively. Vitamin D (both vitamin D2 and D3, calciol) originating from diet or endogenous skin synthesis is delivered to the liver by vitamin D-binding protein (VDBP). There, vitamin D is metabolized by vitamin D 25-hydroxylase (CYP2R1 and CYP27A1) to 25(OH)D (calcidiol), which is the major circulating form of vitamin D in the serum, and its circulating concentration is accepted as an indicator of vitamin D status in a human organism [31,32]. 25(OH)D is further metabolized by 25(OH)D 1α-hydroxylase (CYP27B1) mainly in the proximal tubule of the kidney to 1α,25-dihydroxyvitamin D (1α,25(OH)2D, calcitriol), which is the recognized biologically active form of vitamin D (Figure 1) [31,32]. Calcitriol then enters the circulation and, after binding to VDBP, is delivered to target tissues such as the intestine, bone, and kidney, where vitamin D is known to regulate absorption, mobilization, and reabsorption, respectively, of calcium and phosphate [29]. After being produced, the levels of both calcidiol and calcitriol are tightly regulated by 25(OH)D 24-hydroxylase (CYP24A1), which is the primary vitamin D inactivating enzyme catalyzing hydroxylation at C-24 and C-23 of both calcidiol and calcitriol [31,32]. This 24-hydroxylation pathway produces the biologically inactive calcitroic acid excreted in the bile [33]. The importance of this inactivation step, mediated by CYP24A1, was highlighted in CYP24A1 knockout mice showing impaired intramembranous bone mineralization and hypercalcemia, leading to a lethal phenotype in 50% of the mice [34,35]. However, this defect was rescued in CYP24A1 and VDR double-knockout mice, which suggested that it is the increased calcitriol levels and not the absence of downstream metabolites that were responsible for the flawed phenotype [35].
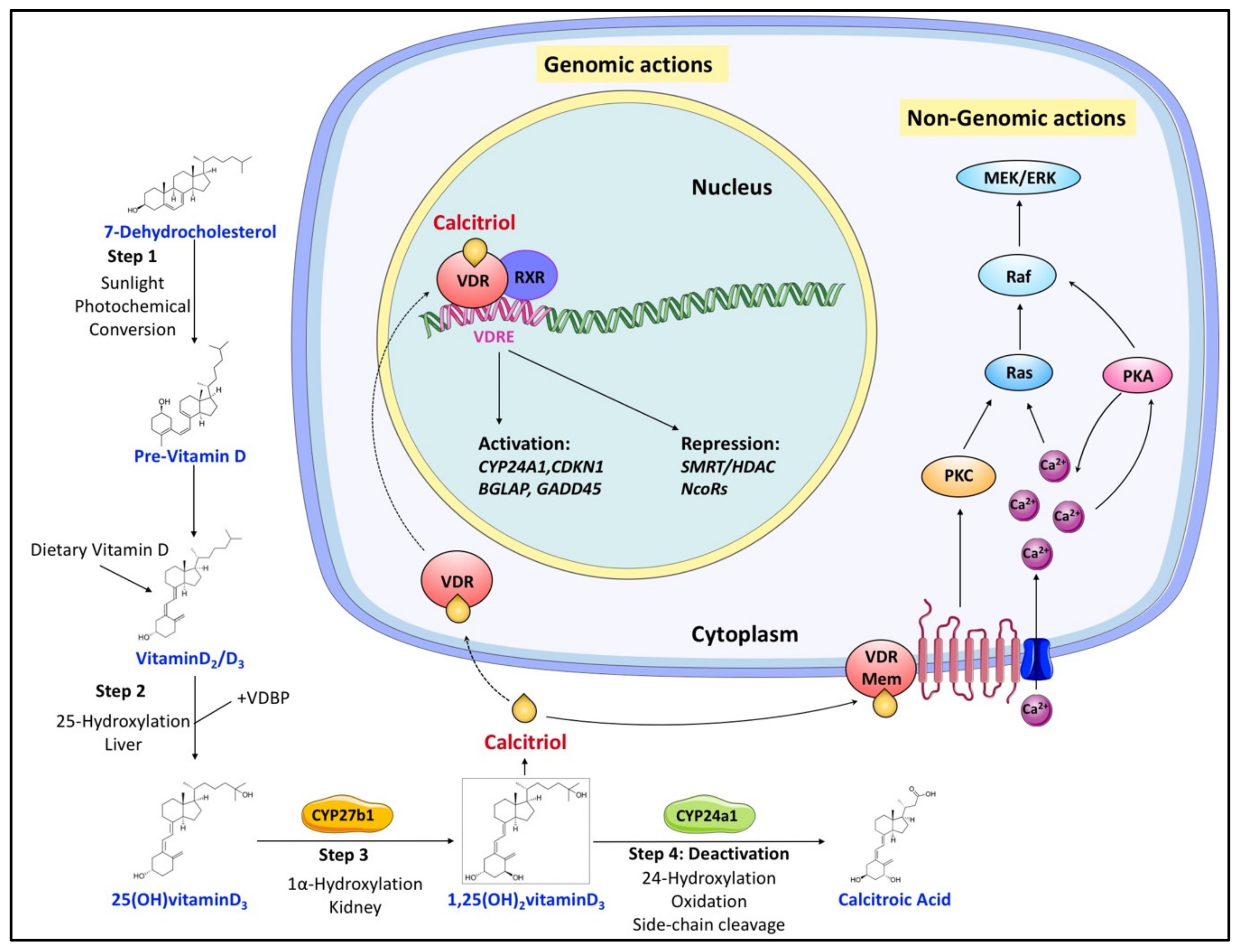
Figure 1. Overview of vitamin D canonical metabolism and its genomic or nongenomic effects. Dietary or cutaneously synthesized vitamin D undergoes two subsequent hydroxylation steps in the liver and kidney to produce active calcitriol (1,25(OH)2 vitaminD3). Calcitriol exerts its functions either by binding to VDR to regulate gene expression or by associating with extracellular binding sites to modulate signaling pathways that influence various cellular processes. Regulation of calcitriol levels also requires inactivation steps mainly involving its hydroxylation by CYP24a1.
2. Noncanonical Vitamin D Metabolic Pathway
Alternatively, vitamin D metabolism is mediated by CYP11A1 (known as a cytochrome P450 side-chain cleavage (P450scc) enzyme) [36]. Vitamin D serves as an alternative substrate for CYP11A1 instead of cholesterol and is sequentially hydroxylated, predominantly at C-20 or C-22, without the cleavage of the side chain producing a multitude of metabolites [37]. Overall, it is estimated that this alternative path produces more than 21 hydroxy-metabolites of vitamin D [36]. Summarily, CYP11A1 products exhibit: (i) antiproliferative, differentiating, and anti-inflammatory abilities in skin cells comparable to that of calcitriol [38,39], (ii) are involved in defense pathways against UVB-induced damage and oxidative stress, and (iii) elicit anticancer abilities in a cell-specific manner [40]. As a point of interest, these alternative metabolites and normal 1,24,25-(OH)3 vitamin D3 do not activate VDR. Thus, the calcemic effects or expression of CYP24A1 can be seen only in response to calcitriol.
3. Hormonal Regulation of the Canonical Vitamin D Metabolic Pathway
As a result of its diverse function, calcitriol is tightly regulated in a negative feedback mechanism [33,41]. Calcitriol inactivation primarily involves modification by CYP24A1, which is among the most prominent targets of the calcitriol–VDR–RXR complex (Figure 1) [42]. In addition, calcitriol can also induce CYP24A1 expression by recruiting histone H4 acetyltransferases and RNA polymerase II to a site approximately 50–70 kb downstream of the human CYP24A1 gene [43]. So, calcitriol signaling levels are tightly kept in control by calcitriol-driven expression of CYP24A1.
Independently, vitamin D metabolism is regulated by two hormones, parathyroid hormone (PTH) and fibroblast growth factor-23 (FGF-23), both of which maintain the calcium and phosphate homeostasis [44]. PTH, secreted by the parathyroid gland in response to calcium levels, stimulates the expression of CYP27B1, leading to an increase in calcitriol production [45]. Although calcitriol signals its degradation via CYP24A1, PTH sustains calcitriol levels by activating the renal cAMP–PKA pathway and invoking the CYP24A1 mRNA degradation [46]. FGF-23, secreted by osteoblasts and osteocytes in response to both phosphate and calcitriol levels [42], reduces serum calcitriol levels by inhibiting the expression of CYP27B1 and simultaneously enhancing the expression of CYP24A1 in the kidney [47].
This entry is adapted from the peer-reviewed paper 10.3390/cancers14071791
This entry is offline, you can click here to edit this entry!