Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Cyclin-dependent kinase 4/6 (CDK4/6) are key regulators of the cell cycle and are deemed as critical therapeutic targets of multiple cancers. Various approaches have been applied to silence CDK4/6 at different levels, i.e., CRISPR to knock out at the DNA level, siRNA to inhibit translation, and drugs that target the protein of interest. Here we summarize the current status in this field, highlighting the mechanisms of small molecular inhibitors treatment and drug resistance.
- CDK4/6
- PROTAC
- small molecular inhibitor
- drug resistance
- cancer
- palbociclib
1. Introduction
Cell division is one of the fundamental biological processes, which functions in various physical and pathological activities [1]. The series of stages sequentially occurring in cell division compose a cell cycle, which includes two successive periods: interphase and mitosis. The former features DNA synthesis in the S phase, before and after which there are G1 and G2 phases, preparing for DNA replication and mitosis, respectively. The latter is marked by sister chromatid segregation. Independent of a cell cycle also exists a dormant G0 phase, where most non-proliferative cells in the human body stay [2].
Normally, the cell cycle is monitored by a quality control system called checkpoints, which consist of G1/S checkpoint (DNA damage checkpoint), G2/M checkpoint, and mitotic spindle checkpoint. These checkpoints detect abnormal cell division and then induce cell cycle arrest, allowing cells to repair defects, thus ensuring proper DNA synthesis and chromosome separation. In addition, whether a cell steps into a cell cycle relies on both extrinsic (e.g., growth factors) and intrinsic (e.g., protein synthesis) signals. The lack of these factors leads cells to enter the G0 phase [1]. The majority of human cells are quiescent, except those in the hematopoietic system or gut epithelium [3].
While in malignant cells, the cell cycle is deregulated, which is characterized by abnormal and uncontrollable cell division. Cancer-related cell cycle defects occur via mutations on multiple proteins essential at different stages of the cell cycle [2]. These genomic dysfunctions dispose of cells to acquire more mutations and numerical aberrations in chromosomes, which reflects abnormal cell division; the accumulated genome mutations result in constitutive mitogenic signals and deficiency of response to anti-mitogenic signals, which brings about unscheduled cell division [4].
The cell cycle has to be tightly regulated owing to its critical role in carcinogenesis. In general, transition through the cell cycle is an orderly process regulated by a series of proteins, of which cyclin-dependent kinases (CDKs) are the most critical ones. CDKs are a family of serine/threonine protein kinases, which form heterodimers with their respective regulatory cyclin subunits [5]. CDKs are generally divided into three groups according to their functions: mitosis-related CDKs (CDK1, CDK2, CDK4, and CDK6), which directly promote cell cycle progression, although there are also other CDKs that work in mammalian cell cycle regulation [4]; transcription-related CDKs (CDK7, CDK8, and CDK9); and atypical CDKs (CDK5, CDK14, CDK15, CDK16, CDK17, and CDK18) [1].
In the cell cycle, CDKs are periodically activated at specific points by cyclins [2]. First, in G1, proliferative signals are sensed by D-type cyclins, which activate CDK4 and CDK6. Subsequently, the expression of E-type cyclins activates CDK2, and the formation of CDK2-cyclin E complex is necessary for G1/S progression. Then in the late S phase, CDK2 is activated by cyclin A, driving transition toward G2. At the end of G2, CDK1-cyclin A complex onsets mitosis. Finally, following nuclear envelope breakdown in prophase, A-type cyclins are degraded, and CDK1-cyclin B promotes cells through mitosis [4]. Besides cyclins, CDKs activity is also regulated by CAK (CDK-activating kinase, i.e., CDK7-cyclin H complex) and CKI (CDK inhibitors), which comprise INK4 (inhibitor of CDK4) proteins and Cip/Kip (CDK-interacting protein/kinase inhibitor protein). Under cell cycle defects, checkpoints can be activated via regulation of CDKs activity and therefore prevent daughter cells from inheriting the faults.
Among various CDKs, CDK4 and CDK6 (CDK4/6) are critical because they play a fundamental role in the G1/S transition. CDKs function via phosphorylating specific substrates [1]. In the G1 phase, the most important substrate for CDK4/6 is retinoblastoma susceptibility protein (Rb), which interacts with the E2F transcriptional family in its hypophosphorylated state, whereby it suppresses transcription of target genes. When a cell senses mitogenic signals, CDK4/6 are activated by cyclin D and then phosphorylate Rb, thus relieving the inhibition [5]. CDK4/6 also contributes to this process by separating Cip/Kip proteins from cyclin E-CDK2 complex, which facilitates CDK2 activation and the following complete phosphorylation of Rb. Sequential phosphorylation of Rb by CDK4/6 and CDK2 results in E2F activation and transcription initiation of genes required for S phase progression (Figure 1). On the other hand, CDK4/6 do not sequester Cip/Kip family under antiproliferative signals; thus, cyclin E-CDK2 is inhibited and cell cycle is arrested at G1 phase.
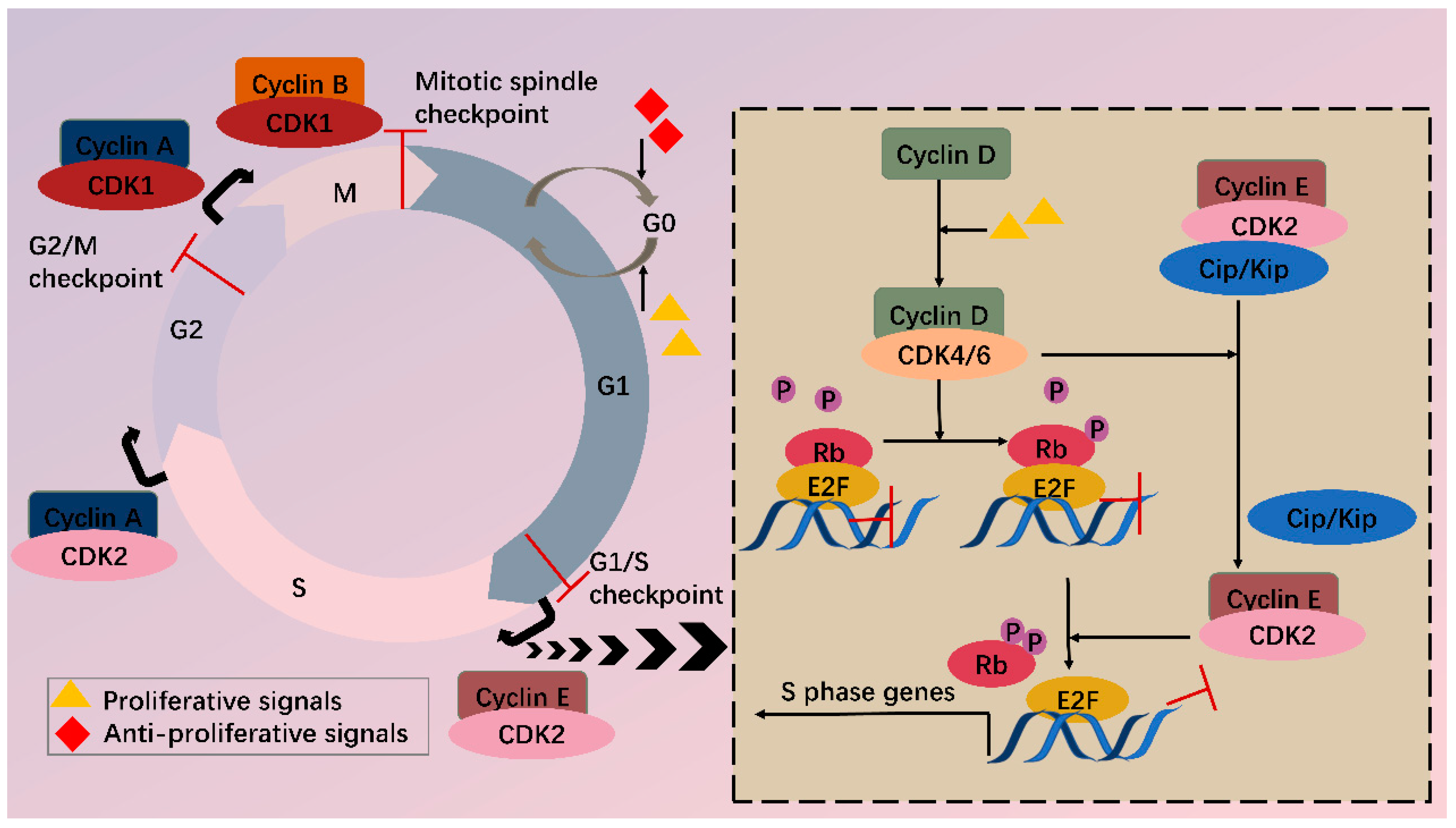
Figure 1. CDKs and cell cycle. Cell cycle consists of G1, S, G2 phase and mitosis. Independent of a cell cycle also exists a dormant G0 phase. Whether a cell steps into a cell cycle relies on balance between proliferative and antiproliferative signals. During a cell cycle, multiple CDKs are sequentially activated. In G1, activated CDK4/6 by cyclin D phosphorylate Rb, partially relieving inhibition of E2F by Rb. Meanwhile, CDK4/6 hijacks Cip/Kip proteins, which stimulate CDK2-cyclin E, facilitating complete phosphorylation of Rb. Thus, E2F activity is totally released, and transcription of S phase genes is initiated. In late G1, CDK2-cyclin E complex is formed, driving transition toward the S phase. Next, CDK2 and CDK1 are successively activated by cyclin A and contribute to S/G2 and G2/M conversion, respectively. Finally, CDK1-cyclin B complex functions during mitosis. Besides CDKs, checkpoints also participate in the cell cycle via regulation of CDKs activity, inducing cell cycle arrest when abnormal cell division is detected.
2. CDK4/6 Are Attractive Targets for Anticancer Treatment
In malignant cells, mutations on and dysregulation of assorted cell cycle regulators, such as CDKs, cyclins, CAK, CKI, CDK substrates, and checkpoint proteins, have been frequently observed [2]. It has been well recognized that the expression levels of CDK4/6 are significantly higher in many tumors [6][7][8][9]. Overexpressed CDK4/6 boost G1/S conversion through directly and indirectly (by stimulating CDK2) phosphorylating Rb, facilitating tumorigenesis. In addition to overexpression, CDK4/6 hyperactivation is common in miscellaneous malignancies (breast cancer, lung cancer, prostate cancer, melanoma, leukemia, lymphoma, glioma, sarcoma, etc.) with differentiated tissue preferences for CDK4/6. CDK4 tends to amplify in epithelial tumors (i.e., in varied cancers) and certain sarcomas, while its homolog favors mesenchymal tissues (including leukemias and sarcomas) [4]. Most human tumors retain wild-type Rb [5], and inhibition of overexpressed or hyperactivated CDK4/6 in these cells can arrest the cell cycle in G1. Even in Rb(-) tumors, CDK4/6 inhibitors also function by reducing cell entry into mitosis or inducing apoptosis in an Rb-independent way [10]. Furthermore, targeting CDK4/6 can also inhibit their cell cycle-independent functions in tumorigenesis. Transcriptomic profiling in breast cancer has uncovered that CDK4 modulates inflammatory cytokine signaling [11]. CDK6 can induce angiogenesis, stem cell activation, immune response, etc. [12].
Moreover, many oncogenes cause cancers by activating the CDK4/6-Rb-E2F pathway and inducing cell proliferation. These include but are not limited to JAK/STAT, PI3K/Akt/mTOR, RAS/RAF/MEK/ERK, BTK/NF-ΚB, and Wnt/β-catenin pathways [13]. In addition, mutations in tumor suppressors such as p53 can also activate the CDK4/6-Rb-E2F pathway by releasing p21CIP1 inhibition. CDK4/6 thus serves as a hub in tumorigenesis pathways. Especially in ER+ breast cancer, there is mutual activation between ER and cyclin D, which will be clarified further (Figure 2). Knockout studies have shown that CDK4/6 are critical for tumor cell growth whereas may be dispensable in normal cells. It is, therefore, safe to kill the enemy without hurting friendly forces [14]. These features together make CDK4/6 appealing and safe targets for anticancer therapy.
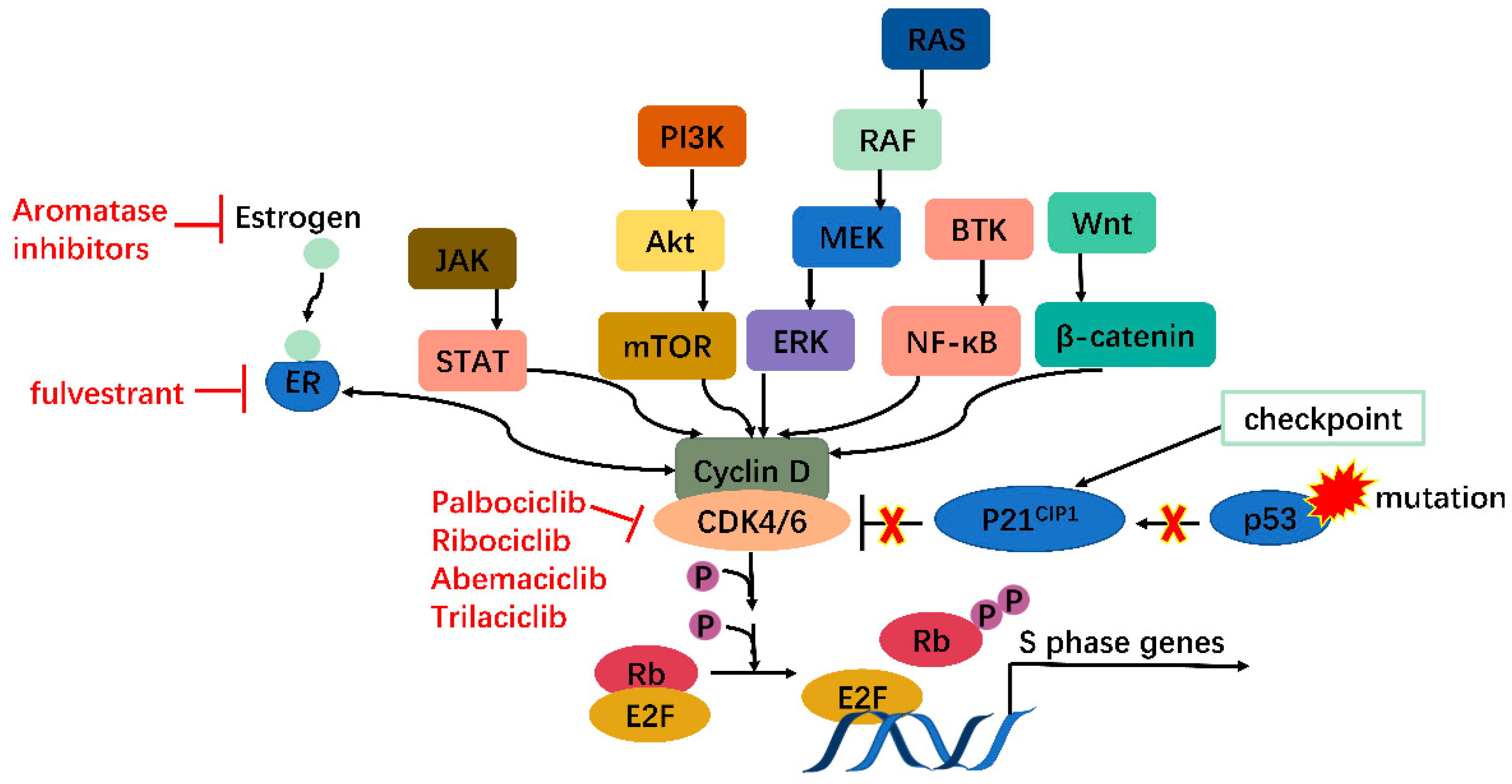
Figure 2. CDK4/6 serves as a hub in tumorigenesis. In cancer, multiple oncogenes may be activated, including those on JAK/STAT, PI3K/Akt/mTOR, RAS/RAF/MEK/ERK, BTK/NF-κB, Wnt/β-catenin pathways, etc., all of which meet at CDK4/6-cyclin D complex. Moreover, mutations on tumor suppressor genes such as p53 can enhance CDK4/6 activity via releasing P21CIP1 inhibition. CDK4/6, therefore, serves as a hub in tumorigenesis.
CDKs, their regulators, and substrates are targets of human cancers. Generally, there are two therapeutic interventions: indirectly targeting CDK regulators or directly focusing on CDKs. The complexity of CDK modulation provides diverse possibilities for indirect tactics, involving lessening or raising the quantity of cyclins or CKI severally, changing activity of those regulators, etc. [2]. Direct policies are attainable at different points of gene expression, which primarily embrace clustered regularly interspaced short palindromic repeats system (CRISPR), RNA interference (RNAi), and protein targeted inhibition or degradation technology. In recent years, direct strategy discovery has been a hot area.
3. CRISPR and RNAi
Silencing CDKs at DNA or RNA level before protein expression is a fascinating approach to cancer therapy. However, very few growth suppression outcomes using CRISPR or RNAi-mediated CDK4/6 or CDK2 deletion have been detected [11][15][16][17]. Contradictorily, there is evidence denying antineoplastic potency of RNAi-based CDK4/6 or CDK2 knockdown [5][18]. In contrast, CRISPR or RNAi treatment targeting other CDKs (CDK7, -8, -9 for CRISPR and CDK1, -10, -12 for RNAi) [19][20][21][22][23] has successfully diminished tumor proliferation.
The disparity between interphase CDKs (CDK2, -4, -6) and CDK1 regarding responsiveness to CRISPR or RNAi is thought-provoking. Among the four CDKs linked to mitosis, CDK1 alone is enough to drive the cell cycle, while CDK2 and CDK4/6 are only indispensable in cell proliferation of specific cells [4][24]. Meanwhile, the lack of cyclin D-CDK4/6 complex can be compensated by cyclin D-CDK1 or cyclin D-CDK2; similarly, cyclin E-CDK 1 or cyclin E-CDK4 can make up for cyclin E-CDK2 absence [5][25][26]; likewise, CDK1 and CDK2 act in a partially redundant way [27]. Hence, the overlapping jobs of mitogenic CDKs and the relatively dispensable position of CDK2 and CDK4/6 compared with CDK1 may partly account for that discrepancy. Namely, loss of function of CDK1 triggered by CRISPR or RNAi cannot be fully offset by interphase CDKs, while it remains possible for CDK1 to fully compensate inhibition of interphase CDKs.
4. Small Molecular Inhibitors
Research on CDK4/6 small molecule inhibitors (SMIs) is an active area. SMIs impede ATP binding to small, determined pockets of CDKs, which holds more potential than hindering large protein-protein interfaces (e.g., interface of cyclin-CDK). However, there are over 500 protein kinases in human genomes, yet ATP binding sites of CDKs are highly conservative [3], so selective drug discovery is challenging. To handle the problem, selectivity determinants outside of the ATP binding pockets are required [28]. The first-generation CDK inhibitors were pan-CDK inhibitors. Due to their poor selectivity and high toxicity, the majority have not been approved for clinical use. The second generation was aimed at improving selectivity for CDK1 and CDK2 and/or total strength [29], and the third generation developed selectivity for CDK4/6 [26]. In addition to poor selectivity and high toxicity, resistance to SMIs cannot be ignored either. Clinical application of CDK4/6 SMIs appears to display weak effects in some tumors (including colorectal cancer, triple-negative breast cancer, melanomas, etc.), where innate or rapid acquisition of resistance may happen [29]. As mentioned, when CDK4/6 are in deficiency, CDK1 or CDK2 can take their roles. Similar events may occur in tumors chronically exposed to CDK4/6 SMIs [5], which may partially explain the mechanisms of resistance, and more reasons will be elucidated hereinafter.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10030685
This entry is offline, you can click here to edit this entry!