Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Mapping non-canonical cellular pathways affected by approved medications can accelerate drug repurposing efforts, which are crucial in situations with a global impact such as the COVID-19 pandemic. Fluoxetine and fluvoxamine are well-established and widely-used antidepressive agents that act as serotonin reuptake inhibitors (SSRI-s). Interestingly, these drugs have been reported earlier to act as lysosomotropic agents, inhibitors of acid sphingomyelinase in the lysosomes, and as ligands of sigma-1 receptors, mechanisms that might be used to fight severe outcomes of COVID-19.
- COVID-19
- SARS-CoV-2
- fluoxetine
- fluvoxamine
- acid sphingomyelinase
- SSRI
- sigma-1 receptors
- FIASMA
- SLC22A3
- lysosomotropic agents
1. Molecular Mechanisms
1.1. Molecular Mechanisms of SARS-CoV-2 Replication and the Involvement of Lysosomes in the Replication Process
SARS-CoV-2 is a beta Coronavirus that is a member of the Coronaviridae family, a large 26.0–32.0 kb enveloped RNA virus with a positive-sense single-stranded RNA genome. The life cycle of SARS-CoV-2 is truly complex with steps that are regulated in both space and time, and, indeed, the researchers currently have adequate information on its structural biology and pathogenesis exemplified by extensive reviews [1][2]; therefore, the researchers are only opting to highlight important steps that are crucial for the cohesion of this manuscript. Briefly, following attachment to host target cell receptors, among which angiotensin-converting enzyme 2 (ACE 2) is considered primary, engagement of the ACE 2 receptors results in a conformational change to the Spike (S) protein, followed by subcleavage by furin and target-cell proteases, such as TMPRSS2 and cathepsin L [3][4].
Entry into the cytoplasm and release of the viral genome is followed by the expression of viral polyproteins that are proteolytically processed into four structural and sixteen non-structural proteins, mediated by the viral papain-like protease (PLpro), the chymotrypsin-like protease (3CLpro) that is also referred to as Main protease (Mpro) [5]. Viral polyproteins are subsequently processed by the viral proteases into 16 non-structural proteins, which are crucial to viral replication and transcription [6]. Additionally, a number of subgenomic mRNAs, nested negative sense RNAs resulting from discontinued transcription of genomic RNA, were found to code for accessory proteins among others, which were linked to host cellular immune responses [7][8][9]. Akin to other Beta Coronaviruses, it is thought that SARS-CoV-2 structural proteins, in addition to viral RNA and other non-structural and accessory proteins, form replication complexes, assembling at sites close to the endoplasmic reticulum (ER) and Golgi compartments characterized by membrane tubules and double-membrane vesicles, possibly derived from the ER [10][11]. Thereafter, viral genomic RNA, along with the nucleocapsid, is thought to translocate to budding sites where other structural glycoproteins are located, followed by assembly and release, utilizing ER trafficking and lysosomal egress [5][12]. It is now apparently clear that the replication cycle of SARS-CoV-2, similarly to other Coronaviruses, heavily relies on the ER, hijacking ER stress responses to facilitate protein translation [13].
1.2. Molecular Mechanisms through Which Fluoxetine and Fluvoxamine Might Prevent the Development of Severe COVID-19
1.2.1. Binding to Sigma-1 Receptors
Fluvoxamine was hypothesized to exert its antiviral effects in the context of COVID-19 through different mechanisms. Of the greatest significance in this process is its interaction with the sigma-1 receptors (S1Rs). S1R is a multifunctional chaperone protein located within the ER that mediates signalling cross-talks in the ER-mitochondria and ER-nucleus context. As a chaperone, it facilitates proper folding of newly synthesized proteins, in addition to preventing the accumulation of misfolded proteins; therefore, playing a major role in cell survival and modulating ER Ca2+ influx into the mitochondria during cellular stress [14]. Under stressful conditions, i.e., viral infection, the overexpression of S1Rs plays a protective role, reducing ER and oxidative stress, counteracting pro-apoptotic signals through the induction of a wide range of mechanisms that promote cell survival [15].
Given their pivotal role in mitigating cellular stress during a viral infection, S1Rs ligands are frequently explored as potential drugs against viral infections and are currently being studied against SARS-CoV-2. For example, in the context of HCV infection, S1Rs were found to colocalize with non-structural proteins associated with the viral replicase complex [16], and more recently, the nsp6 of SARS-CoV-2 was found to directly interact with S1R, highjacking cellular translation machinery, thus, favouring expression of viral proteins. Molecules targeting sigma receptors such as antipsychotic drugs (haloperidol), and antihistamines (clemastine and cloperastine), all of which are sigma receptor ligands, were found to also exert anti-SARS-CoV-2 activity [17]. Fluoxetine, among other antidepressant drugs, was found to perturb the replication of HCV in an unbiased screening cell culture assay [18]. Additionally, the unfolded protein response (UPR) and autophagy are also ER stress responses that are exploited by SARS-CoV-2, and the S protein in β-Coronaviruses was found to modulate UPR, favouring viral protein synthesis and replication [19][20]. Through its modulatory effect on the ER stress response, fluvoxamine may therefore hinder the replication of SARS-CoV-2 [21].
1.2.2. Lysosomal Membrane Composition as the Potential Site of Action of Fluoxetine and Fluvoxamine in COVID-19
Decreasing the acidity of endosomal pH adversely affects endosomal function and trafficking and inhibits the activity of the endosomal proteases [22]. Modulation of endolysosomal pH could potentially impair the formation of viral replication complexes, in addition to impeding viral trafficking and budding [23]. Both fluoxetine and fluvoxamine, among other SSRIs, are considered to be lysosomotropic agents, and hence, it is plausible that the lysosomotropic and endolysosomal pH-modulating effects of these molecules may indeed show potential beneficial effects in the context of COVID-19.
The role of acid sphingomyelinase (ASM) in augmenting viral infection has long been known and described. Covering a wide range of cellular functions, from cytoskeletal reorganization to proliferation, response to stress, signalling, and induction of apoptosis, acid sphingomyelinase catalyses the hydrolysis of sphingomyelin to phosphorylcholine and ceramide. Being a predominantly lysosomal enzyme, proteolytic activation of the ASM can result in its translocation to the cellular membrane and the generation of ceramide at the extracellular cell surface [24][25]. Ceramide is an integral part of the cellular membrane, modulating its biophysical properties, and is composed of a sphingosine backbone that can be post-translationally modified, yielding complex molecules, including glycosphingolipids (GSL). Although there has been documented evidence of the involvement of ceramide and ceramide-based molecules in mediating several viral infections, such as Rotavirus, HIV, and Influenza A, it is unclear whether ceramide or one of its derivatives are involved in mediating the attachment or internalization of virion particles [26][27][28]. In regards to SARS-CoV-2, the main receptor (ACE 2) was found to locate in lipid rafts [29] and the effect of the ASM inhibition, namely the change in the ceramide content of the lipid rafts, was found to inhibit viral infection of the cells in in vitro studies [30]. SSRIs such as fluoxetine or fluvoxamine were found to accumulate in lysosomes and disturb and attenuate the activity of ASM [31]. Indeed, fluoxetine treatment, through this proposed mechanism, efficiently inhibited the infection of Vero-E6 cell lines with SARS-CoV-2 and vesicular stomatitis virus pseudo viral particles enveloped with SARS-CoV-2 S protein [30][32].
1.2.3. Anti-Inflammatory Effect of SSRI-s
Some of the potentially relevant SSRI antidepressants may also exert their protective effect against severe COVID-19 through the down-regulation of the inflammatory response induced by a viral infection, independently of their action on the S1Rs. The possible mechanism of action proposed was through modulating the immune activity of the macrophages [33], while depression itself is considered by some to be an inflammatory disease [34]. The role of macrophages in the anti-inflammatory activity of antidepressant drugs has been reviewed thoroughly and recently by Nazimek et al. [35].
In severe COVID-19, a state of platelet dysregulation is observed, marked by increased activation, reactivity, and aggregation [36][37]. Both fluoxetine and fluvoxamine block the re-uptake of serotonin from plasma in platelets via the sodium-dependent serotonin transporter (SERT), and by limiting the uptake of serotonin, SSRIs interfere with platelet activation and aggregation, therefore, potentially increasing bleeding time and reducing neutrophil recruitment and inflammation [38]. It is therefore conceivable that SSRIs may be of some benefit in advanced COVID-19, counteracting the hypercoagulable state of platelets observed. The topic is still controversial [39], but based on clinical studies, fluoxetine appears to prevent platelet activation and is more likely to prevent thrombotic events [40].
In an in-vivo multiple sclerosis (MS) rat model, fluvoxamine treatment resulted in a significant increase in the viability and proliferation of neural stem cells, and treatment with physiological concentrations attenuated the severity of encephalomyelitis, manifested by a decrease in serum levels of IFN-γ, and an increase in IL-4, pro-, and anti-inflammatory cytokines, respectively [41].
2. Experimental Data That Support the Concept That Fluoxetine Might Be Useful in Treating COVID-19
Several specific experimental investigations have been performed during the COVID-19 pandemic that underlie the possibility that fluoxetine might be beneficial in the fight against the development of severe outcomes of COVID-19 infection.
2.1. Experimental Data from the Ursula Rensher Group
In a paper accepted in September 2020, before vaccines became available, the group of Ursula Rensher from Münster (Germany) reported a series of experiments in which they had investigated molecules that interfered with cholesterol accumulation in late endosomes [32].
The model cell lines were Vero E6 cells and polarized bronchial Calu-3 cells. The line of Vero cells was established in 1962 from the kidneys of a normal African green monkey. They are non-tumorigenic cells, widely used in vaccine production in standardized conditions [42]. VeroE6 cells are widely used in experiments that study the pathomechanisms of SARS-CoV-2 infection as they can be easily infected with the virus and the released virus particles from these cells can be easily and precisely quantitated by real-time QPCR measurements [43]. While Vero E6 cells are excellent for assessing viral replication, the Calu-3 cells established in 1975 from a pleural effusion of lung adenocarcinoma [44] are good models for epithelial lung cell modelling.
The authors used fluoxetine, imipramine, amiodarone, and the NPC1 inhibitor U18666A. NPC1 is an intracellular cholesterol transport protein that transports low-density lipoproteins into the late endosomes, mutation of NPC1 can cause Niemann Pick type C disease. As they could show that fluoxetine was active against viral replication for influenza virus strains (EC50 = 1 µM and EC90 = 5–6 µM), they tested its effect on SARS-CoV-2 cells and found that EC90 for SARS-CoV-2 was in the range of 2–4 µM depending on the used cell lines. U18666A could reduce viral titers by 99% at the concentration of 10 µM-s. Imipramine and amiodarone were also effective on viral replication in different cell lines.
The authors investigated cholesterol accumulation in late endosomes using microscopic methods. As a positive control, they used U18666A treated cells. They could show a significant accumulation of cholesterol at a very high dose of fluoxetine of 20 µM-s, but at a lower dose of 5 µM-s the results were not significant. The authors tested the changes in the pH of the endosomes by microscopic methods. In this assay, both 5 µM and 20 µM-s of fluoxetine produced significant changes in lysosomal pH, comparable to those caused by 2–10 µM-s of U18666A.
Finally, they compared the infectivity of Vero E6 cells pre-treated with 5 µM fluoxetine or 10 µM of U18666A. Both pre-treatments produced a significant reduction of infectivity, as assessed by intracellular nucleocapsid detection with microscopic methods.
In their next study, accepted for publication in February 2021, at the time when global vaccination campaigns had just been launched, the same group published a manuscript where they argued that remdesivir, itraconazole, and fluoxetine might have synergistic effects on SARS-CoV-2 infection in vitro [45]. They used the same cell lines as in their previous report. The concentration of fluoxetine used in their study was between 0.5 µM and 2.5 µM. At the highest dose of 2.5 µM fluoxetine concentration, without remdesivir co-treatment, the virus titer reduction was negligible. Although they could show synergies for both the combination of remdesivir with Itraconazole and the combination of remdesivir with fluoxetine, a potential limitation of their experimental work was that remdesivir had a very short half-life in serum. In mice experiments, remdesivir was detectable in serum only half an hour after dosing. It is to be noted that the potentially active metabolites of remdesivir could be detected in serum up to 24 h post-dosing [46]. At the same time, an observation that might underlie the potential synergy between remdesivir and fluoxetine in the lung is, that in the same investigation, the concentration of remdesivir in lung tissue could be detected much longer, up to 24 h with the highest concentration at two hours. The reported concentration of remdesivir in the lung tissue was 0.35 µM. At a similar concentration of remdesivir (0.25 µM) in the Renscher study, 2.5 µM of fluoxetine produced only a modest 10% reduction in the virus titer.
In September 2021 the Reschler group published a study [47] wherein they investigated the synergy of fluoxetine with the GS-441524 nucleoside analogue. GS-441524 is the main plasma metabolite of remdesivir with a plasma half-life of 24 h. Human data on GS-441524 are scarce, the current applications are mainly experimental and veterinary [48]. In this paper, the Rescher group investigated the synergy between various doses of fluoxetine and GS-441524. The same Vero E6 and Calu-3 cells were used. Similar synergies were observed as for remdesivir. The highest used concentration was 2.5 µM of fluoxetine, which had marginal inhibitory effects as seen in their previous report. A combination of 2.5 µM fluoxetine with 1 µM of GS-441524 produced a 99.9% inhibition of virus production on polarized Calu-3 cells.
2.2. Experimental Data from the Jochen Bodem Group
In March 2021, the group of Jochen Bodem published a short but important paper [49]. Their results confirmed the findings of the Rescher group, namely that fluoxetine dramatically reduced the viral replication of SARS-CoV-2 in the Vero E6 cell line that originates from the kidneys of a normal African green monkey. Similarly to the results of the Rensher group, in the experiments of the Bodem group, the concentration of 2.5 µM of fluoxetine resulted in the reduction of virus titer by one order of magnitude, approx. 90% inhibition, moreover, 5 µM of fluoxetine had a dramatic effect of more than three orders of magnitude reduction of the viral titer. At these concentrations of fluoxetine, no significant inhibition of cell growth was seen in Vero6 cells. Escitalopram or Paroxetine had marginal effects. The results were quantified by virus-specific QPCR and confirmed by microscopic staining. Fluoxetine did not affect other tested viruses such as RSV, Rabies, HSV-1, and HHV8.
The same paper by Bodem [49] reports a very important experiment assessing the effect of fluoxetine on viral replication in normal human lung tissue preparations. Human, disease-free lung tissue slices of 300 µm width with intact peripheral airways were prepared and cultivated from samples originating from lobe resection due to cancer. The tissue slices were treated with 5 µM of fluoxetine and then infected with SARS-CoV-2. After 3 days, supernatants were harvested, and infectivity was assessed in Vero E6 cells. The resulting virus titers were quantified by QPCR. Fluoxetine treatment of the lung slices at 5 µM-s resulted in a more than two orders of magnitude reduction of viral output in the developed assay, which corresponds to more than 99% of inhibition. These fluoxetine concentrations are in line with the concentrations measured in postmortem human lung samples as described later.
2.3. Enantiomer Indifference of the Antiviral effect of Fluoxetine
A third very important observation is reported in the same paper of Bodem [49] that addresses the stereoselectivity of fluoxetine effects. The currently used fluoxetine is the racemic mixture of both S and R enantiomers. In their report, the authors investigated the viral replication inhibitory effect of both the racemic mixture and the two enantiomers separately. In their experiments they found that there was no difference in the inhibitory effects on the virus between the two enantiomers. This observation is of extreme importance if the researchers take into account the previous knowledge about the specifically psychiatric effects of the two fluoxetine enantiomers, which was accumulated in the 1990s. Although the enantiomers of fluoxetine were not studied extensively, the enantiomers of norfluoxetine, the metabolic product generated by demethylation in the liver and which are also active serotonin reuptake inhibitors, have been well investigated. The topic was examined in several studies assessing rat brains and complemented with studies on human platelets [50][51]. These investigations showed that the S enantiomer of norfluoxetine was over 20 fold more potent than the R enantiomer regarding the SSRI effect of the enantiomers. Interestingly, the less effective R enantiomer, if administered orally at 80 or 120 mg/day, resulted in QT elongations on ECG measurements. These QT elongations were statistically significant, underlying the possibility that the cardiac effects of fluoxetine observed in other studies [52] might involve other mechanisms beyond SSRI activity. It is to be noted that the QT elongation effect of fluoxetine, although reported, is still less pronounced compared to the similar effect of citalopram [53], where these cardiac side effects led to the development of escitalopram, which contains the S enantiomer of citalopram to reduce potential side effects. The observations on the similar inhibitory effect on SARS-CoV-2 replication of both S and R fluoxetine enantiomers, together with the 20 fold higher SSRI effect of the S enantiomer, raise the possibility of the development of an antiviral formulation based on the R enantiomer that might have fewer CNS effects. As this is the first report on the enantiomer indifference of fluoxetine antiviral effects, further studies are needed to confirm these observations.
3. Pharmacokinetics of Fluoxetine and Fluvoxamine
3.1. Pharmacokinetic Data
In order to assess the opportunity for using fluoxetine or fluvoxamine in blunting the severity of the effects of COVID-19, the researchers need to investigate the pharmacological context of their administration. The two molecules present a quite similar pharmacokinetic profile. As the published literature focuses more on fluoxetine, the researchers will first present the data available about fluoxetine and at the end of this sub-chapter the researchers will provide an overview of the major differences regarding fluvoxamine.
Fluoxetine was introduced into clinical practice in 1987 and since then has been widely used in different concentrations and for a relatively wide spectrum of indications in the field of psychiatric perturbances or altered states [54][55].
A particularly beneficial feature of fluoxetine is that several physiological or pathological states that might need an adjustment of the therapeutic dose do not influence its tissue distribution and local concentrations. Fluoxetine is well absorbed, has a significant first-pass hepatic metabolism and low lipid solubility, and as a result, age, obesity, or renal failure do not dramatically affect fluoxetine pharmacokinetics [56][57]. At the same time, in case of liver impairment, e.g., in alcoholic liver cirrhosis, the pharmacokinetics changed significantly, resulting in a more than two-fold increase in the half-life of the molecule [58].
Another great practical feature of fluoxetine is that its bioavailability is not affected by food. A single dose of oral administration will result in a peak plasma level after 6–8 h, with a maximal CNS efficacy as assessed by EEG, detected after 8–10 h post oral administration. In the liver, a demethylation step occurs that generates Norfluoxetine, which is also active as an inhibitor of serotonin reuptake. The significant first-pass metabolism in the liver that generates norfluoxetine can lead to a single dose of oral fluoxetine of 40 mg-s in different persons achieving different maximal plasma concentrations [56][57] ranging from 15 µg/L to 55 µg/L, which corresponds to 50–150 nanomolar (nM) plasma concentrations. If 60 mgs were administered daily, the steady-state plasma level was achieved relatively late, after 30 days, and 80% was excreted in the urine, while 15% in faeces (with 5% of the radiocarbon used for tracing not found in either urine or faeces). Initially, the recommended dose was 80 mg/day, but later 20 mg/day was shown to be more effective. Moreover, in the elderly age group, 3 times 20 mg weekly was shown to be equally effective.
The pharmacokinetics of fluvoxamine have been well described and summarized in several reviews [55][59]. Fluvoxamine is well and rapidly absorbed after oral administration, reaching a maximum plasma concentration level somewhat earlier than fluoxetine. While the range of tmax for fluoxetine is considered to be 6–8 h, for fluvoxamine these values are between 2 and 8 h. After oral administration, 96% of fluvoxamine is absorbed, which is a relatively higher fraction compared to fluoxetine, where 80% is absorbed. Both drugs have a strong first-pass hepatic metabolism. As for excretion, 4% of fluvoxamine and its metabolites are excreted with urine while only 80% of fluoxetine and its metabolites are eliminated through the kidneys, and the rest is eliminated with the faeces. Pharmacokinetics of fluvoxamine are not significantly affected by the status of the kidney, but hepatic cirrhosis does affect the elimination of both drugs. Age does not seem to affect the metabolism of any of these two molecules significantly, so long as therapeutic doses are followed.
3.2. Oral Dose versus Body Fluid Concentrations
In order to be able to compare the results of the performed clinical studies and reported experimental data, the researchers need to compare the achieved tissue concentrations and the concentrations used in the different experiments. For performing such a comparison, the researchers need to address the issue that cellular experiments report their concentrations in micromoles (µM), while clinical dosage and tissue concentrations are usually expressed in micrograms (µg) and microgram per gram or microgram per millilitre. The molecular weight of fluoxetine is 309.33 g/mol. The currently used typical dose of 20 mgs of fluoxetine is equivalent to 64.65 nanomoles. Tissue distribution is little affected by the amount of adipose tissue in the patient. Peak plasma concentrations after a single dose of 30–40 mg are in the range of 15–55 µg/L corresponding to 48–177 nano Mols. As for tissue distributions, interestingly, the highest concentration of the drug was found in the lung and liver in early measurements performed in dogs and confirmed in human samples as well [54]. In brain tissue, the levels of fluoxetine seem to be two-fold higher (average 2.6 fold higher in some cases 4.9 fold higher) compared to plasma levels, with significant patient-to-patient variability [60][61].
In a post-mortem assessment of body fluids collected from deceased pilots after aviation fatalities [62], the concentration of fluoxetine from all investigated tissues was the highest in lung tissue samples, exceeding 60 fold the concentration in blood with significant person-to-person variability. Similar values were found for the degradation product norfluoxetine, in which case the concentration in the lung was 59 times higher in the lung tissue compared to blood concentration. The highest fluoxetine concentration was 51.9 µg/g in the lung tissue, with a mean of 19.6 µg/g, these concentrations are in the range of 50 µM concentration, with the lowest measured concentration of 1.56 µg/g corresponding to 5 µM. The measurements from post-mortem lung specimens resulted in drug levels in the same range, or even higher than the concentrations of 2–10 µM-s used in the published cell assays that were investigating the effect of fluoxetine on SARS-CoV-2 virus replication in various cell lines. The concentrations in plasma in pharmacologic studies were in the range of 100 nM. The postmortem data show 60-fold enrichment in lung tissues, relative to blood, and based on this, the researchers can conclude that the lung concentration is very likely to be in the range of 5 µM, which is similar to the effective concentration reported in cellular assays.
Some lung-specific side effect reports in the early 1990s underlie the possibility that fluoxetine might specifically target the lung. Pulmonary or systemic phospholipidosis caused by fluoxetine was documented in animals and reported in humans [63], while direct fluoxetine-induced lung damage without a known cause was reported several times in early case reports [64][65]. These observations underline the enrichment of fluoxetine and its derivatives in lung tissues that might be of extreme importance in preventing severe effects of SARS-CoV-2 infection.
3.3. Tissue Distribution of Fluoxetine Drug Transporters
In order to explain the surprisingly high concentration in the lung and the specific effects reported earlier, the researchers investigated what is known in the literature regarding the transporters responsible for the transport of fluoxetine into different body compartments. Fluoxetine is transported by the protein Organic Cation Transporter 3 (OCT-3), which has the canonical name of SLC22A3. A review on various aspects of organic cation transporters and their involvement in the transport of various drugs in different tissues was published recently [66]. Based on the public human tissue-specific gene expression database of the Human Proteome Atlas, on a single-cell level [67] the highest expression of the gene is seen on alveolar cells type 2, followed by hepatocytes and pancreatic endocrine cells, which for hepatocytes, is in concordance with the strong first-pass hepatic metabolism. Regarding the lung, this might explain the high tissue concentration reported earlier. Interestingly, an observation underlying the transport in pancreatic cells is that fluoxetine-induced beta-cell dysfunction was also reported in cellular systems [68][69] in animal studies [70], and a systematic review performed based on Danish patient registries reported a significantly increased risk of pancreatitis in first-time users of SSRIs [71].
3.4. The Potential Target Cells in the Alveolae Responsible for the Protective Effect of Fluoxetine and Fluvoxamine in SARS-CoV-2 Infections
As reported during the first waves of the pandemic, the most severe outcome of COVID-19 disease was the progressive patient deterioration approximately a week after the onset of symptoms with a decrease in oxygen saturation levels and progressive decompensation of the respiratory system.
In order to understand the relationship between the two SSRI molecules investigated and the progression of COVID-19 disease, the researchers will focus on the cellular distribution on the alveolar level of key molecules involved in viral infection and SSRI activity.
Based on the public human tissue-specific gene expression database of the Human Proteome Atlas, on the single-cell level [67], in the lung non-vascular cells, the highest expression of ACE2, the most likely cellular entry point of the SARS-CoV-2 virus, is expressed only on alveolar type 2 cells and the expression level is marginal [72].
Interestingly, in this single-cell-level assessment of RNA levels from the different types of cells present in the alveolae, the highest expression level of SLC22A3 was present, on the type 2 alveolar epithelial cells, while the other cells of the alveolae, namely the alveolar epithelial cells type I, macrophages, club cells, fibroblasts, ciliated cells, and other immune cells of the alveolae had a significantly lower or marginal expression [73].
What are type 2 alveolar cells and what is their importance? As described by Robert J. Mason [74], at stage 3 of the COVID-19 disease, hypoxia and ground-glass infiltrates develop with a progression towards acute respiratory distress syndrome (ARDS). At this stage, the virus reaches the gas exchange units or alveolae and infects preferentially type 2 alveolar cells similarly to the influenza virus. Infected type 2 cells release further viruses, contribute to the infection of nearby alveolae, and later undergo apoptosis. The same author in a later article [75] explains relevant characteristics of type 2 alveolar cells. These cells are involved in keeping the alveolae fluid-free through their specific expression of CFTR chloride ion transporter. Type 1 alveolar cells contribute to maintaining the fluid-free state of the alveolae through CLIC5, another chloride ion transporter. These statements are confirmed by single-cell RNA seq data as presented here [73] and here [76].
Type 2 alveolar cells have various functions. In Figure 1 the researchers present the distinctive role of the type 2 alveolar cells and their role in surfactant production and SARS-CoV-2 virus replication. They can be considered the stem cells of the alveolae [77] that can both regenerate themselves and also give rise to the type 1 alveolar cells, those flat cells that cover the largest part of the alveolae. Their overall differentiation takes up to one year. Another major function of these cells is that they are the ones producing the surfactant, a mixture of different lipids that cover the alveolae that is essential in keeping them open during respiration. The surfactant is a mixture of various lipids stored in the lamellar bodies of type 2 cells, organelles that can be considered as modified lysosomes. These lamellar bodies release their content by exocytosis. The released surfactant is in part degraded by macrophages or a smaller part is recycled by type 2 alveolar cells [78]. The critical connection between surfactant metabolism and lysosomal storage diseases was described in detail by Tamara Paget, Emma J. Parkinson-Lawrence, and Sandra Orgeig [79]. The apoptosis of the viral infected type 2 cells could affect the respiratory function in many ways. With the decrease in surfactant production, the alveolae could collapse, and as a result of the diminished chloride ion transport, fluids might accumulate in the alveolae, and by the destruction of the stem cell pool of the alveolae, the long-term regenerative potential of the lung might be diminished. As a conclusion, the researchers can state that type 2 alveolar cells are critical in maintaining the physiological conditions of the lung, they are targets of the SARS-CoV-2 virus and express the transporter of fluoxetine, therefore they are excellent candidates for the protective effects of fluoxetine in case of COVID-19.
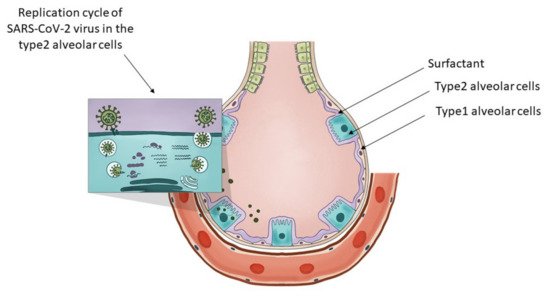
Figure 1. The key role of type 2 alveolar cells in the surfactant production and SARS-CoV-2 infection at the level of the lung alveolae. The insert on the top-left presents the major steps of the replication cycle of SARS-CoV-2 virus in the type 2 alveolar cells.
3.5. Mechanisms for Lysosome Enrichment of Fluoxetine
In addition to a likely high intensity of the transport of fluoxetine by OCT3/SLC22A3 into alveolar cells, other mechanisms might lead to high levels in the lung, such as (1) phospholipid binding and (2) lysosomal trapping [80]. The experiments that suggest these mechanisms were performed in different types of rat tissue slices immersed in media containing fluoxetine. From a medium containing 5 µM of fluoxetine, the accumulation ratio was highest in the lung tissue slices (75.6%) and moderate accumulation (10.5%) was observed on brain tissue slices. This accumulation was reduced by roughly a quarter by adding lysosomal inhibitors such as monensin or NH4Cl. Similar accumulation in the lung was seen for promazine, imipramine, amitriptyline, sertraline, and carbamazepine, too. In this early study, as a mechanism for the lung accumulation of the drug in lung tissues, it was suggested that the abundance of lysosomes in the lung alveolar macrophages, together with the abundance of surfactant rich in phospholipids, might be responsible. At the same time, although the brain does contain a high amount of phospholipids, it showed only moderate accumulation of these drugs. According to the early hypothesis by Korhuber [81], the slow accumulation of lysosomotropic psychoactive agents in the brain might contribute to the delayed effects observed in clinical practice, and may disturb several biochemical processes that require an acidic milieu, such as the proton-driven transport of monoamines into synaptic vesicles. As such, the accumulation of fluoxetine as a lysosomotropic agent in lung tissue might dramatically change the function of lysosomes.
The hypothesis that lysosomotropic agents could be beneficial in fighting COVID-19 was elaborated in detail in several papers, including one by Homolak in June 2020 [12] where some clinical studies involving lysosomotropic agents were also listed. The topic was elaborated in the same month by Blaess [82] as part of potential new therapeutic strategies, where a list of proposed lysosomotropic agents was presented. One of the proposed representatives of the lysosomotropic agents was Azithromycin. This antibiotic was tested in the early phases of the COVID-19 pandemic for preventing severe symptoms, but no statistically significant results could be shown [83]. Chloroquine and hydroxychloroquine, also lysosomotropic agents, were widely investigated for preventing severe outcomes of COVID-19. Currently, these drugs are not recommended for the treatment of COVID-19 in combination with Azithromycin, as a large body of evidence, such as the RECOVERY trial [60], the Solidarity trial [84], and the PETAL trial [85], proved their ineffectiveness in COVID-19. The NIH COVID-19 treatment guidelines as of early 2022 do not recommend these drugs to be used to prevent severe COVID-19 outcomes [86]. These observations are against the hypothesis that lysosomotropic agents, in general, could help prevent severe COVID-19 outcomes, and suggest that other mechanisms might be responsible for the potentially beneficial effects observed in the case of fluoxetine and fluvoxamine in the clinical studies presented.
References
- Gusev, E.; Sarapultsev, A.; Solomatina, L.; Chereshnev, V. SARS-CoV-2-specific immune response and the pathogenesis of COVID-19. Int. J. Mol. Sci. 2022, 23, 1716.
- Nile, S.H.; Nile, A.; Qiu, J.; Li, L.; Jia, X.; Kai, G. COVID-19: Pathogenesis, cytokine storm and therapeutic potential of interferons. Cytokine Growth Factor Rev. 2020, 53, 66–70.
- Peacock, T.P.; Goldhill, D.H.; Zhou, J.; Baillon, L.; Frise, R.; Swann, O.C.; Kugathasan, R.; Penn, R.; Brown, J.C.; Sanchez-David, R.Y.; et al. The furin cleavage site in the SARS-CoV-2 spike protein is required for transmission in ferrets. Nat. Microbiol. 2021, 6, 899–909.
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 2020, 181, 271–280.e8.
- V’Kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus biology and replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170.
- Chen, Y.; Liu, Q.; Guo, D. Emerging coronaviruses: Genome structure, replication, and pathogenesis. J. Med. Virol. 2020, 92, 2249.
- Yang, H.; Rao, Z. Structural biology of SARS-CoV-2 and implications for therapeutic development. Nat. Rev. Microbiol. 2021, 19, 685–700.
- Masters, P.S. Coronavirus genomic RNA packaging. Virology 2019, 537, 198–207.
- Wang, D.; Jiang, A.; Feng, J.; Li, G.; Guo, D.; Sajid, M.; Wu, K.; Zhang, Q.; Ponty, Y.; Will, S.; et al. The SARS-CoV-2 subgenome landscape and its novel regulatory features. Mol. Cell 2021, 81, 2135–2147.e5.
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martinez-Sobrido, L.; Garcia-Sastre, A.; Weber, F.; Kochs, G. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 2007, 361, 304–315.
- Oudshoorn, D.; Rijs, K.; Limpens, R.; Groen, K.; Koster, A.J.; Snijder, E.J.; Kikkert, M.; Barcena, M. Expression and cleavage of middle east respiratory syndrome coronavirus nsp3-4 polyprotein induce the formation of double-membrane vesicles that mimic those associated with coronaviral RNA replication. MBio 2017, 8, e01658-17.
- Homolak, J.; Kodvanj, I. Widely available lysosome targeting agents should be considered as potential therapy for COVID-19. Int. J. Antimicrob. Agents 2020, 56, 106044.
- Sola, I.; Almazan, F.; Zuniga, S.; Enjuanes, L. Continuous and discontinuous RNA synthesis in coronaviruses. Ann. Rev. Virol. 2015, 2, 265–288.
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell 2007, 131, 596–610.
- Vasallo, C.; Gastaminza, P. Cellular stress responses in hepatitis C virus infection: Mastering a two-edged sword. Virus Res. 2015, 209, 100–117.
- Friesland, M.; Mingorance, L.; Chung, J.; Chisari, F.V.; Gastaminza, P. Sigma-1 receptor regulates early steps of viral RNA replication at the onset of hepatitis C virus infection. J. Virol. 2013, 87, 6377.
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468.
- Mingorance, L.; Friesland, M.; Coto-Llerena, M.; Perez-del-Pulgar, S.; Boix, L.; Lopez-Oliva, J.M.; Bruix, J.; Forns, X.; Gastaminza, P. Selective inhibition of hepatitis C virus infection by hydroxyzine and benztropine. Antimicrob. Agents Chemother. 2014, 58, 3451–3460.
- Fung, T.S.; Liao, Y.; Liu, D.X. Regulation of stress responses and translational control by coronavirus. Viruses 2016, 8, 184.
- Chan, C.P.; Siu, K.L.; Chin, K.T.; Yuen, K.Y.; Zheng, B.; Jin, D.Y. Modulation of the unfolded protein response by the severe acute respiratory syndrome coronavirus spike protein. J. Virol. 2006, 80, 9279–9287.
- Sukhatme, V.P.; Reiersen, A.M.; Vayttaden, S.J.; Sukhatme, V.V. Fluvoxamine: A review of its mechanism of action and its role in COVID-19. Front. Pharmacol. 2021, 12, 652688.
- Shu, X.; Sun, Y.; Sun, X.; Zhou, Y.; Bian, Y.; Shu, Z.; Ding, J.; Lu, M.; Hu, G. The effect of fluoxetine on astrocyte autophagy flux and injured mitochondria clearance in a mouse model of depression. Cell Death Dis. 2019, 10, 577.
- Prasad, H.; Prasad, C.H. Protons to patients: Targeting endosomal Na+/H+ exchangers against COVID-19 and other viral diseases. FEBS J. 2021, 288, 5071–5088.
- Henry, B.; Ziobro, R.; Becker, K.A.; Kolesnick, R.; Gulbins, E. Acid sphingomyelinase. Handb. Exp. Pharmacol. 2013, 215, 77–88.
- Beckmann, N.; Becker, K.A. Ceramide and related molecules in viral infections. Int. J. Mol. Sci. 2021, 22, 5676.
- Martinez, M.A.; Lopez, S.; Arias, C.F.; Isa, P. Gangliosides have a functional role during rotavirus cell entry. J. Virol. 2013, 87, 1115–1122.
- Magerus-Chatinet, A.; Yu, H.; Garcia, S.; Ducloux, E.; Terris, B.; Bomsel, M. Galactosyl ceramide expressed on dendritic cells can mediate HIV-1 transfer from monocyte derived dendritic cells to autologous T cells. Virology 2007, 362, 67–74.
- Suzuki, Y.; Matsunaga, M.; Matsumoto, M. N-Acetylneuraminyllactosylceramide, GM3-NeuAc, a new influenza A virus receptor which mediates the adsorption-fusion process of viral infection. Binding specificity of influenza virus A/Aichi/2/68 (H3N2) to membrane-associated GM3 with different molecular. J. Biol. Chem. 1985, 260, 1362–1365.
- Lu, Y.; Liu, D.X.; Tam, J.P. Lipid rafts are involved in SARS-CoV entry into Vero E6 cells. Biochem. Biophys. Res. Commun. 2008, 369, 344–349.
- Carpinteiro, A.; Edwards, M.J.; Hoffmann, M.; Kochs, G.; Gripp, B.; Weigang, S.; Adams, C.; Carpinteiro, E.; Gulbins, A.; Keitsch, S.; et al. Pharmacological inhibition of acid sphingomyelinase prevents uptake of SARS-CoV-2 by epithelial cells. Cell Rep. Med. 2020, 1, 100142.
- Breiden, B.; Sandhoff, K. Emerging mechanisms of drug-induced phospholipidosis. Biol. Chem. 2019, 401, 31–46.
- Schloer, S.; Brunotte, L.; Goretzko, J.; Mecate-Zambrano, A.; Korthals, N.; Gerke, V.; Ludwig, S.; Rescher, U. Targeting the endolysosomal host-SARS-CoV-2 interface by clinically licensed functional inhibitors of acid sphingomyelinase (FIASMA) including the antidepressant fluoxetine. Emerg. Microbes Infect. 2020, 9, 2245–2255.
- Nazimek, K.; Kozlowski, M.; Bryniarski, P.; Strobel, S.; Bryk, A.; Myszka, M.; Tyszka, A.; Kuszmiersz, P.; Nowakowski, J.; Filipczak-Bryniarska, I. Repeatedly administered antidepressant drugs modulate humoral and cellular immune response in mice through action on macrophages. Exp. Biol. Med. 2016, 241, 1540–1550.
- Maes, M. Depression is an inflammatory disease, but cell-mediated immune activation is the key component of depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 664–675.
- Nazimek, K.; Strobel, S.; Bryniarski, P.; Kozlowski, M.; Filipczak-Bryniarska, I.; Bryniarski, K. The role of macrophages in anti-inflammatory activity of antidepressant drugs. Immunobiology 2017, 222, 823–830.
- Manne, B.K.; Denorme, F.; Middleton, E.A.; Portier, I.; Rowley, J.W.; Stubben, C.; Petrey, A.C.; Tolley, N.D.; Guo, L.; Cody, M.; et al. Platelet gene expression and function in patients with COVID-19. Blood 2020, 136, 1317–1329.
- Hottz, E.D.; Azevedo-Quintanilha, I.G.; Palhinha, L.; Teixeira, L.; Barreto, E.A.; Pao, C.R.R.; Righy, C.; Franco, S.; Souza, T.M.L.; Kurtz, P.; et al. Platelet activation and platelet-monocyte aggregate formation trigger tissue factor expression in patients with severe COVID-19. Blood 2020, 136, 1330–1341.
- Duerschmied, D.; Suidan, G.L.; Demers, M.; Herr, N.; Carbo, C.; Brill, A.; Cifuni, S.M.; Mauler, M.; Cicko, S.; Bader, M.; et al. Platelet serotonin promotes the recruitment of neutrophils to sites of acute inflammation in mice. Blood 2013, 121, 1008–1015.
- Serebruany, V.L.; Suckow, R.F.; Cooper, T.B.; O’Connor, C.M.; Malinin, A.I.; Krishnan, K.R.R.; van Zyl, L.T.; Lekht, V.; Glassman, A.H. Relationship between release of platelet/endothelial biomarkers and plasma levels of sertraline and N-desmethylsertraline in acute coronary syndrome patients receiving SSRI treatment for depression. Am. J. Psychiatry 2005, 162, 1165–1170.
- Wozniak, G.; Toska, A.; Saridi, M.; Mouzas, O. Serotonin reuptake inhibitor antidepressants (SSRIs) against atherosclerosis. Med. Sci. Monit. 2011, 17, RA205.
- Ghareghani, M.; Zibara, K.; Sadeghi, H.; Dokoohaki, S.; Sadeghi, H.; Aryanpour, R.; Ghanbari, A. Fluvoxamine stimulates oligodendrogenesis of cultured neural stem cells and attenuates inflammation and demyelination in an animal model of multiple sclerosis. Sci. Rep. 2017, 7, 4923.
- Barrett, P.N.; Mundt, W.; Kistner, O.; Howard, M.K. Vero cell platform in vaccine production: Moving towards cell culture-based viral vaccines. Expert Rev. Vaccines 2014, 8, 607–618.
- Xian, H.; Liu, Y.; Rundberg Nilsson, A.; Gatchalian, R.; Crother, T.R.; Tourtellotte, W.G.; Zhang, Y.; Aleman-Muench, G.R.; Lewis, G.; Chen, W.; et al. Metformin inhibition of mitochondrial ATP and DNA synthesis abrogates NLRP3 inflammasome activation and pulmonary inflammation. Immunity 2021, 54, 1463.
- Memorial Sloan Kettering Cancer Center. Calu-3: Human Lung Cancer Cell Line. Available online: https://www.mskcc.org/research-advantage/support/technology/tangible-material/calu-3-human-lung-cell-line (accessed on 21 January 2022).
- Schloer, S.; Brunotte, L.; Mecate-Zambrano, A.; Zheng, S.; Tang, J.; Ludwig, S.; Rescher, U. Drug synergy of combinatory treatment with remdesivir and the repurposed drugs fluoxetine and itraconazole effectively impairs SARS-CoV-2 infection in vitro. Br. J. Pharmacol. 2021, 178, 2339–2350.
- Hu, W.-J.; Chang, L.; Yang, Y.; Wang, X.; Xie, Y.-C.; Shen, J.-S.; Tan, B.; Liu, J. Pharmacokinetics and tissue distribution of remdesivir and its metabolites nucleotide monophosphate, nucleotide triphosphate, and nucleoside in mice. Acta Pharmacol. Sin. 2020, 42, 1195–1200.
- Brunotte, L.; Zheng, S.; Mecate-Zambrano, A.; Tang, J.; Ludwig, S.; Rescher, U.; Schloer, S. Combination therapy with fluoxetine and the nucleoside analog GS-441524 exerts synergistic antiviral effects against different SARS-CoV-2 variants in vitro. Pharmaceutics 2021, 13, 1400.
- Amirian, E.S.; Levy, J.K. Current knowledge about the antivirals remdesivir (GS-5734) and GS-441524 as therapeutic options for coronaviruses. One Health 2020, 9, 100128.
- Zimniak, M.; Kirschner, L.; Hilpert, H.; Geiger, N.; Danov, O.; Oberwinkler, H.; Steinke, M.; Sewald, K.; Seibel, J.; Bodem, J. The serotonin reuptake inhibitor fluoxetine inhibits SARS-CoV-2 in human lung tissue. Sci. Rep. 2021, 11, 5890.
- Fuller, R.W.; Snoddy, H.D.; Krushinski, J.H.; Robertson, D.W. Comparison of norfluoxetine enantiomers as serotonin uptake inhibitors in vivo. Neuropharmacology 1992, 31, 997–1000.
- Wong, T.; Bymaster, F.P.; Reid, L.R.; Mayle, D.A.; Krushinski, J.H.; Robertson, D.W. Norfluoxetine enantiomers as inhibitors of serotonin uptake in rat brain. Neuropsychopharmacology 1993, 8, 337–344.
- Varriale, P. Fluoxetine (prozac) as a cause of QT prolongation. Arch. Intern. Med. 2001, 161, 612.
- Funk, K.A.; Bostwick, J.R. A comparison of the risk of QT prolongation among SSRIs. Ann. Pharmacother. 2013, 47, 1330–1341.
- Benfield, P.; Heel, R.C.; Lewis, S.P. Fluoxetine. Drugs 2012, 32, 481–508.
- Hiemke, C.; Härtter, S. Pharmacokinetics of selective serotonin reuptake inhibitors. Pharmacol. Ther. 2000, 85, 11–28.
- Altamura, A.C.; Moro, A.R.; Percudani, M. Clinical pharmacokinetics of fluoxetine. Clin. Pharmacokinet. 1994, 26, 201–214.
- Aronoff, G.R.; Bergstrom, R.F.; Pottratz, S.T.; Sloan, R.S.; Wolen, R.L.; Lemberger, L. Fluoxetine kinetics and protein binding in normal and impaired renal function. Clin. Pharmacol. Ther. 1984, 36, 138–144.
- Schenker, S.; Bergstrom, R.F.; Wolen, R.L.; Lemberger, L. Fluoxetine disposition and elimination in cirrhosis. Clin. Pharmacol. Ther. 1988, 44, 353–359.
- Van Harten, J. Clinical pharmacokinetics of selective serotonin reuptake inhibitors. Clin. Pharmacokinet. 1993, 24, 203–220.
- RECOVERY Collaborative Group. Effect of hydroxychloroquine in hospitalized patients with Covid-19. N. Engl. J. Med. 2020, 383, 2030–2040.
- Renshaw, P.F.; Guimaraes, A.R.; Fava, M.; Rosenbaum, J.F.; Pearlman, J.D.; Flood, J.G.; Puopolo, P.R.; Clancy, K.; Gonzalez, R.G. Accumulation of fluoxetine and norfluoxetine in human brain during therapeutic administration. Am. J. Psychiatry 1992, 149, 1592–1594.
- Johnson, R.D.; Lewis, R.J.; Angier, M.K. The distribution of fluoxetine in human fluids and tissues. J. Anal. Toxicol. 2007, 31, 409–414.
- Gonzalez-Rothi, R.; Zander, D.S.; Ros, P.R. Fluoxetine hydrochloride (prozac)-induced pulmonary disease. Chest 1995, 107, 1763–1765.
- Davies, L.P. Comment on Fluoxetine-induced lung damage. Med. J. Aust. 1992, 156, 740.
- Bass, S.P.; Colebatch, H.J.H. Fluoxetine-induced lung damage. Med. J. Aust. 1992, 156, 364–365.
- Samodelov, S.L.; Kullak-Ublick, G.A.; Gai, Z.; Visentin, M. Organic cation transporters in human physiology, pharmacology, and toxicology. Int. J. Mol. Sci. 2020, 21, 7890.
- Karlsson, M.; Zhang, C.; Méar, L.; Zhong, W.; Digre, A.; Katona, B.; Sjöstedt, E.; Butler, L.; Odeberg, J.; Dusart, P.; et al. A single–cell type transcriptomics map of human tissues. Sci. Adv. 2021, 7, eabh2169.
- Chang, H.Y.; Chen, S.L.; Shen, M.R.; Kung, M.L.; Chuang, L.M.; Chen, Y.W. Selective serotonin reuptake inhibitor, fluoxetine, impairs E-cadherin-mediated cell adhesion and alters calcium homeostasis in pancreatic beta cells. Sci. Rep. 2017, 7, 3515.
- Elmorsy, E.; Al-Ghafari, A.; Helaly, A.N.M.; Hisab, A.S.; Oehrle, B.; Smith, P.A. Editor’s highlight: Therapeutic concentrations of antidepressants inhibit pancreatic beta-cell function via mitochondrial complex inhibition. Toxicol. Sci. 2017, 158, 286–301.
- Youssef, S. Effect of fluoxetine on the pancreas of adult male albino rats and the possible protective role of omega-3: Light and electron microscopic study. Int. J. Clin. Dev. Anat. 2017, 3, 45.
- Nørgaard, M.; Jacobsen, J.; Gasse, C.; Pedersen, L.; Mortensen, P.B.; Sørensen, H.T. Selective serotonin reuptake inhibitors and risk of acute pancreatitis: A population-based case-control study. J. Clin. Psychopharmacol. 2007, 27, 259–262.
- Single Cell Type—ACE2—The Human Protein Atlas. Available online: https://web.archive.org/web/20220215154750/https://www.proteinatlas.org/ENSG00000130234-ACE2/single+cell+type/lung (accessed on 15 February 2022).
- Wayback Machine. Available online: https://web.archive.org/web/20220215155450/http://web.archive.org/screenshot/https://www.proteinatlas.org/ENSG00000146477-SLC22A3/single+cell+type/lung (accessed on 15 February 2022).
- Mason, R.J. Pathogenesis of COVID-19 from a cell biology perspective. Eur. Respir. J. 2020, 55, 2006607.
- Mason, R.J. Thoughts on the alveolar phase of COVID-19. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L115–L120.
- Single Cell Type—CLIC5—The Human Protein Atlas. Available online: https://web.archive.org/web/20220215161636/https://www.proteinatlas.org/ENSG00000112782-CLIC5/single+cell+type/lung (accessed on 28 February 2022).
- Barkauskas, C.E.; Cronce, M.J.; Rackley, C.R.; Bowie, E.J.; Keene, D.R.; Stripp, B.R.; Randell, S.H.; Noble, P.W.; Hogan, B.L.M. Type 2 alveolar cells are stem cells in adult lung. J. Clin. Investig. 2013, 123, 3025.
- Agudelo, C.W.; Samaha, G.; Garcia-Arcos, I. Alveolar lipids in pulmonary disease. A review. Lipids Health Dis. 2020, 19, 122.
- Paget, T.L.; Parkinson-Lawrence, E.J.; Orgeig, S. Interstitial lung disease and surfactant dysfunction as a secondary manifestation of disease: Insights from lysosomal storage disorders. Drug Discov. Today Dis. Model. 2019, 29–30, 35–42.
- Daniel, W.A.; Wójcikowski, J. Contribution of lysosomal trapping to the total tissue uptake of psychotropic drugs. Pharmacol. Toxicol. 1997, 80, 62–68.
- Kornhuber, J.; Retz, W.; Riederer, P. Slow accumulation of psychotropic substances in the human brain. Relationship to therapeutic latency of neuroleptic and antidepressant drugs? J. Neural. Transm. Suppl. 1995, 46, 315–323.
- Blaess, M.; Kaiser, L.; Sauer, M.; Csuk, R.; Deigner, H.P. COVID-19/SARS-CoV-2 infection: Lysosomes and lysosomotropism implicate new treatment strategies and personal risks. Int. J. Mol. Sci. 2020, 21, 4953.
- Hinks, T.S.C.; Cureton, L.; Knight, R.; Wang, A.; Cane, J.L.; Barber, V.S.; Black, J.; Dutton, S.J.; Melhorn, J.; Jabeen, M.; et al. Azithromycin versus standard care in patients with mild-to-moderate COVID-19 (ATOMIC2): An open-label, randomised trial. Lancet Respir. Med. 2021, 9, 1130–1140.
- WHO Solidarity Trial Consortium. Repurposed antiviral drugs for COVID-19—Interim WHO solidarity trial results. N. Engl. J. Med. 2021, 384, 497–511.
- Self, W.H.; Semler, M.W.; Leither, L.M.; Casey, J.D.; Angus, D.C.; Brower, R.G.; Steven, Y.C.; Collins, S.P.; Eppensteiner, J.C.; Filbin, M.R.; et al. Effect of hydroxychloroquine on clinical status at 14 days in hospitalized patients with COVID-19: A randomized clinical trial. JAMA 2020, 324, 2165–2176.
- Chloroquine or Hydroxychloroquine. COVID-19 Treatment Guidelines. Available online: https://www.covid19treatmentguidelines.nih.gov/therapies/antiviral-therapy/chloroquine-or-hydroxychloroquine-and-or-azithromycin/ (accessed on 16 January 2022).
This entry is offline, you can click here to edit this entry!