Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
The term “autophagy”, (from the Greek words auto, meaning “self” and phagein, meaning “to eat”)—literally, eating one’s self—was first created by Christian de Duve over 40 years ago, who discovered lysosomes and provided clear proof of their participation in this process. It is an evolutionarily conserved process of degradation and recycling in eukaryotic organisms. H2S is already considered a physiological mediator involved in many physiological and pathological processes in animals and plants, including autophagy.
- autophagy
- hydrogen sulfide
- persulfidation
- PTM
1. Introduction
The term “autophagy”, (from the Greek words auto, meaning “self” and phagein, meaning “to eat”)—literally, eating one’s self—was first created by Christian de Duve over 40 years ago, who discovered lysosomes and provided clear proof of their participation in this process [1]. It is an evolutionarily conserved process of degradation and recycling in eukaryotic organisms. Two common forms of autophagy have been described in mammals and plants: micro-autophagy and macro-autophagy, while they differ in a third type of autophagy described, chaperone-mediated autophagy (in mammals) and mega-autophagy (in plants) [2,3,4,5,6]. The differences among them have been previously described in detail elsewhere [7,8], and this review will focus on macro-autophagy (hereafter, autophagy). In this latest process, the cytoplasm and/or organelles are isolated in double membrane vesicles—named autophagosomes—and then transported to the lytic organelle (vacuole in plants and yeast, and lysosome in animals) to be degraded, resulting in the turnover of cellular components. Therefore, autophagy is a fundamental cell clearance pathway that eliminates cellular components, including nucleic acids, proteins, lipids, and organelles, to promote homeostasis, differentiation, development and cell survival.
Autophagy is a unique membrane trafficking process that involves the de novo formation of a membrane, which is generally derived from the endoplasmic reticulum (ER) by generating a double membrane structure called phagophore that elongates to sequester cytoplasmic cargo and closes to form the autophagosome [6,9].
The molecular process of autophagy was mostly unknown until 1993, when Yoshinori Oshumi described a genetic screen in yeast, leading to the discovery of AuTophagy-related Genes (ATG) [10]. 41 yeast ATG genes have been described, and many of them have orthologues in other organisms such as humans and plants.
The autophagy core process in mammals is induced in response to stress by inhibiting the mammalian kinase target of rapamycin (mTOR) or activating 5’ AMP-activated protein kinase (AMPK). In mammals, different stress stimuli can trigger autophagy, such as protein misfolding, hypoxia, nutritional and energy deficiency, ER stress, redox stress, mitochondrial damage, and pathogen infection [11]. Dysregulated autophagy plays an important role in many pathological processes, including ischemia-reperfusion injury, inflammatory and infectious diseases, obesity and type 2 diabetes, cancer and neurodegenerative diseases [12,13,14].
In plants, inhibition of TOR, usually induced by starvation of nutrients such as nitrogen starvation, is the main pathway that triggers autophagy. In addition, it can also be regulated by repression of glucose signaling, activating the energy sensor Snf1-related protein kinase 1 (SnRK1), which in turn inhibits TOR and activates the ATG1 autophagy initiation complex. The function of AMPK in plant autophagy remains largely unknown, although a plant ortholog of mammalian AMPK, named KIN10, was described as a positive regulator of plant autophagy [15]. In plant cells, autophagy is triggered by different biotic and abiotic stresses such as oxidative stress, salinity, hypoxia, heat and cold, nutrient starvation, ER stress and pathogen infection. Therefore, autophagy is essential for plants during reproductive and vegetative development, senescence, starvation, immune response and it is critical to cope with environmental stress [3,16,17]. Thus, autophagy must be tightly regulated to maintain cellular homeostasis.
Hydrogen sulfide (H2S) is a colorless, flammable and highly toxic gas known for its rotten egg scent at low concentrations. It has always been considered a toxic pollutant that is found naturally in sewers, stagnant or well waters, compost pits, gas wells and volcanoes. However, it is also endogenously produced in cells by different enzymes.
H2S is produced in animals by cystathionine β synthase (CBS, EC 4.2.1.22), cystathionine-γ-lyase (CSE, EC 4.4.1.1) and 3-mercaptopyruvate sulfurtransferase (3-MST, EC 2.8.1.2); these use cysteine or 3-mercaptopyruvate as substrates. The sulfate-reducing bacterial flora in the large intestine of animals also releases H2S, reaching concentrations from 0.3 to 3.4 mmol L−1 in the colon [18,19].
In Arabidopsis, the plant species where the H2S signaling has been deeply studied, H2S is produced from cysteine by the action of L-cysteine desulfhydrases (DES1, EC 4.4.1.2; and L-CDES, EC 4.4.1.1), D-cysteine desulfhydrases (D-CDES, EC 4.4.1.15), cyanoalanine synthase (CAS, EC 4.4.1.9), cysteine synthase (CS, EC 4.2.99.8), NifS-like proteins and in the photosynthetic sulfate assimilation pathway by sulfite reductase (SiR, EC 1.8.7.1) [20].
Over the last decade, both in animal and plant systems, H2S has been highlighted as a biological signaling molecule—namely, gasotransmitter—as important as other signal molecules such as nitric oxide (NO), carbon monoxide (CO) or hydrogen peroxide (H2O2) [21,22,23].
H2S is already considered a physiological mediator involved in many physiological and pathological processes in animals and plants. Its regulatory function in mammals includes processes such as reducing inflammation [24], synaptic transmission [25], apoptosis [26], vascular tone [27], ischemia-reperfusion injury [28] and promoting ulcer healing [29] and protects cells from oxidative stress [30]. In plants, H2S regulates a wide range of physiological processes, from seed germination to fruit maturation and the first description of its influence on vegetative development and disease resistance of plants dates from the late 1960s [31,32]. Today, the protective effects of H2S against different stresses are widely known, such as drought [33], osmotic and saline stresses [34], heat [35], oxidative stress [36] and metal stresses [37]. In addition, H2S regulates photosynthesis [38], stomatal closure/aperture [39,40] and autophagy [41,42,43,44,45,46,47].
2. Hydrogen Sulfide as a Regulator of Autophagy
2.1. The Anti-Autophagic Role of Sulfide in Plants
Over the last 10 years, there have been many studies on the effects of H2S on autophagy in eukaryotic cells, but its mechanism has not been completely deciphered.
In plants, the role of H2S in autophagy has been described as a protective effect towards a prosurvival outcome. By now, H2S has been revealed as a negative regulator of autophagy induced by nutrient deficiency, carbon and nitrogen deprivation [41,43] and osmotic [46] and ER stress [45] (Figure 1). It was shown that only sulfide donor molecules, and no other compounds containing inorganic sulfur, are responsible for the inhibition of autophagy under nitrogen starvation in Arabidopsis roots [43]. Besides, sulfide signaling was dose-dependent, with an optimal NaHS (commonly used as sulfide generating molecule) concentration of 100–200 µM, with devastating effects at higher concentrations, inducing autophagy, probably due to its toxicity.
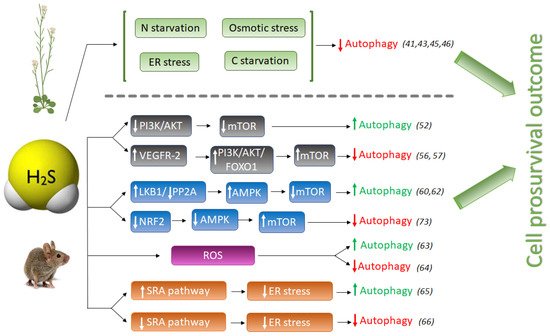
Figure 1. Schematic comparison of pro-autophagic and anti-autophagic effect of H2S signaling in animal and plant systems. H2S, hydrogen sulfide; LKB1, liver kinase B1; PP2A, Protein phosphatase 2; AMPK, adenosine monophosphate-activated protein kinase; mTOR, mammalian target of rapamycin; NRF2, Nuclear factor E2-related factor 2; Akt (PKB), protein kinase B; PI3K, phosphatidylinositol 3-kinase; FOXO1, Forkhead Box O1; VEGFR-2, Vascular endothelial growth factor receptor 2; SRA, Scavenger receptor A. Numbers between brackets refer to references cited.
The induction of autophagy by oxidative stress, especially during nutrient deprivation, and the ability of H2S to activate the antioxidant response of plant cells, are well known. However, it was nicely shown that the negative regulation of autophagy through sulfide signaling was not dependent on its antioxidant activity, showing that hydrogen sulfide does not behave as an H2O2 or superoxide scavenger [43]. Furthermore, treatments with identical concentrations of antioxidant molecules such as glutathione and ascorbate were unable to produce the negative effect that sulfide treatment had on autophagy regulation; only NaHS treatment significantly inhibited autophagy [43].
Hydrogen sulfide has also been revealed to play a key role in autophagy during ER stress. Aggregation of misfolded proteins in the endoplasmic reticulum disrupts ER function, producing ER stress [48], which interferes with normal physiological functions of the cell. ER stress occurs when an increase of misfolded proteins accumulate in the ER which may be activated by different adverse environmental conditions such as cold or heat and pathogen infections in plants [49], or by several chemical and physiological situations such as glucose deprivation, hypoxia or genome instability in animals [50].
In a recent study, the effect of sulfide was also demonstrated to be independent from the antioxidant activity under ER stress, comparing the results observed using similar concentrations of sulfide, glutathione and ascorbate. Their results showed that when ER stress was induced with tunicamycin, no significant decrease in autophagosomes was detected upon well-established antioxidant compounds. By contrast, sulfide provoked a severe decrease of autophagosomes, indicating that the negative effect of sulfide is independent of redox conditions [45].
The anti-autophagic role of sulfide in Arabidopsis was also demonstrated under induced autophagy by carbon starvation, where Cys-generated sulfide in the cytosol was shown to regulate negatively autophagy and to modulate the transcriptional profile of Arabidopsis [41]. DES1, the L-Cys desulfhydrase protein located in the cytosol, catalyzes the desulfuration of L-Cys to sulfide plus ammonia and pyruvate. Consequently, the null mutant des1-1 plants contain 30% less endogenous sulfide in leaves than WT plants. Mutant des1-1 plants were shown to have induced autophagy under physiological conditions, and exogenous treatment with NaHS negatively regulated autophagy in this mutant background [41]. Moreover, sulfide was able to suppress autophagy induction caused by carbon starvation even in wild-type plants, whereas exogenous ammonia, also a product of DES1 activity, had no effect on carbon-induced autophagy. Therefore, it was concluded that sulfide exerts a general effect on autophagy unrelated to nutrition limitation stress.
In a different study sought to decipher the mechanism of action by which NaHS regulates autophagy, it was shown that abscisic acid (ABA) treatment induced the autophagic flux and that this induction was also repressed by NaHS [46]. One of the first plant responses to adverse environmental conditions is the increase of intracellular ABA content in order to activate downstream ABA-signaling pathway so as to help plants cope with the stress. In this situation, when plants successfully have overcome the adverse conditions and induced autophagy is not more required, NaHS repression prevents the over-activation of autophagy allowing to return back to levels in favorable growth conditions [44]. Therefore, in all studies reported up to now in plant systems, sulfide has an anti-autophagic role (Figure 1).
2.2. The Pro- or Anti-Autophagic Role of Sulfide in Mammals
However, the pro- or anti-autophagic role of H2S in mammals has not always been completely clear, and several publications have shown that autophagy and H2S could be a double-edged sword in cancer studies depending on the experimental settings. Hydrogen sulfide induces autophagy of hepatocellular carcinoma cells (HCC) by inhibiting the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (AKT)/ mTOR (PI3K/Akt/mTOR) signaling pathway [51]. PI3Ks are a family of lipid kinases, which phosphorylate phosphoinositides that entail AKT recruitment to the cell membrane. AKT is an evolutionarily highly conserved serine/threonine protein kinase, considered one of the key downstream proteins of PI3K. mTOR is a conserved serine/threonine protein kinase and it is the catalytic core of two complexes: mTORC1 and mTORC2. Activation of the PI3K/Akt pathway further phosphorylates downstream regulators such as mTOR and the transcription factor Forkhead box O-1 (FoxO-1), upregulating the activity of mTOR complex 1 (mTORC1) that drives autophagy inhibition [52]. The PI3K/AKT signaling pathway is one of the upstream pathways that regulate mTOR. Suppression or dysfunction of PI3K can greatly block the downstream signaling pathways AKT and mTOR, and therefore the induction of autophagy (Figure 1).
NaHS treatment significantly inhibited the expression of phospho-PI3K, phospho-Akt, and mTOR proteins in HCC cells, mimicking the effect of rapamycin [51], and therefore activating autophagy. However, NaHS did not affect basal-level Akt phosphorylation in heart disease during ischemia, but further doubled myocardial Akt phosphorylation during reperfusion [53]. Zhou Y. et al., also found that NaHS enhances mTOR phosphorylation in both ischemic and reperfused hearts [53]. In another study, pretreatment of MC3T3-E1 osteoblasts with SDSS [a H2S donor derived from β-(3,4-dihydroxyphenyl)lactic acid] stimulated Akt phosphorylation in a concentration-dependent manner [54]. H2S also activates vascular endothelial growth factor 2 (VEGFR-2), which, in turn, activates the PI3K/AKT/FOXO-1 signaling pathway, with the opposite result of inhibition of autophagy [55,56] (Figure 1).
Thus, the role of NaHS activating the phosphorylation or dephosphorylation of the PI3K/AKT signaling pathway and the outcome of autophagy regulation has not been clearly deciphered in mammals. The different cell types, as well as the sulfide concentrations used in the experiments, were likely the consequence of different conclusions.
The adenosine monophosphate-activated protein kinase (AMPK) is involved in the regulation of metabolic energy balance, and several studies have implicated the AMPK/mTOR pathway in the regulation of autophagy. Numerous publications described the role of H2S in activating autophagy through the AMPK/mTOR pathway, making this signaling a promising target for several diseases [18,57,58].
Another pro-autophagic effect of H2S has been described in the regulation of the liver kinase B1 (LKB1)-AMPK signaling pathway [59]. LKB1 forms a heterotrimeric complex with the pseudokinase Ste20-related adaptor (STRAD) and the scaffolding mouse protein 25 (MO25), and this LKB1-STRAD-MO25 complex activates AMPK by phosphorylation [60]. Kundu et al. described that NaHS treatment in hyperglycemic cells increased LKB1/STRAD/MO25 complex assembly and therefore, AMPK phosphorylation, promoting autophagy [59] (Figure 1). However, a later study demonstrated that H2S regulated AMPK phosphorylation through inhibition of protein phosphatase 2A (PP2A), and not through the LKB1/STRAD/MO25 complex [61], but sulfide still had a pro-autophagic role.
The protective effect of sulfide in several illnesses has been linked to its role promoting autophagy which may decrease ROS production. In a recent study in endothelial progenitor cells (EPCs), exogenous H2S ameliorated the high glucose (HG)-induced injury by promoting autophagic flux and decreasing ROS production, demonstrating the protecting role of sulfide under this dysfunction [62]. Their findings demonstrated that the phosphorylation of the endothelial nitric oxide synthase (eNOS) at Thr495 determines whether this enzyme produces either NO or superoxide, and sulfide reduced the phosphorylation level of this enzyme, decreasing NO and ROS production [62].
But on the other hand, it is well known that oxidative stress may induce autophagy to protect cells from apoptosis. The effect of sulfide ameliorating oxidative stress has also been described in mice where it was demonstrated that GYY4137, a sulfide donor, attenuated the severity of lung injury by alleviating septicemia-induced ferroptosis and inhibiting the activation of autophagy in sepsis-induced acute lung injury [63]. Therefore, sulfide may play an anti-autophagic role by alleviating oxidative stress (Figure 1).
Prolonged ER stress has been associated with a wide range of diseases, including neurodegeneration, cancer, atherosclerosis, type 2 diabetes and liver disease. Autophagy is activated to remove dysfunctional proteins during ER stress. Several studies have connected the role of sulfide enhancing autophagy in reducing ER stress in mammals. In peritoneal macrophages of rats, hydrogen sulfide induces autophagy by suppressing the class A scavenger receptor (SRA) pathway (Figure 1). This cell response reduces ER-stress by inducing autophagy and protects against ischemia/reperfusion injury, maintaining liver function [64]. In other studies, NaHS treatment blocked ER stress and ER stress-associated autophagy [65].
During ER stress, H2S has been reported to inhibit protein tyrosine phosphatase (PTP1B) [66]. PTP1B dephosphorylates PKR-like endoplasmic reticulum kinase (PERK), an ER stress sensor that autophosphorylates and induces the phosphorylation of eukaryotic initiation factor 2 alpha (eIF2α), which is necessary to mediate the induction of autophagy [67]. Therefore, it was concluded that exogenous H2S, or induction of its endogenous synthesis, suppress the activation of PERK/eIF2α/ATF4 pathway and its subsequent effects on ER stress, which are an increased eIF2α phosphorylation [68], and the induction of autophagy. In addition, H2S suppresses the expression of PKR-like endoplasmic reticulum kinase (PERK) [69], which induces autophagy.
Additionally, H2S induces the activity of the transient receptor potential channel (TPRV4) and KATP channels, mediating angiogenesis and inducing vasodilation [70,71]. Through activation of TPRV4, H2S also activates the AMPK/mTOR pathway, by this means reducing autophagy [72].
Hydrogen sulfide also exerts a cytoprotective role by upregulating cellular antioxidants by suppressing nuclear factor erythroid-2 related factor 2 (NRF2) [73]. NRF2 is a family of nuclear basic leucine zipper transcription factors that regulate the gene expression of a number of antioxidant enzymes. However, Nrf2 can also sense ROS and RNS in stressed cells, triggering the activation of AMPK, which suppresses mTOR and therefore induces autophagy [74]. Thus, NaHS could also inhibit excessive autophagy of vascular endothelial cells by the Nrf2/AMPK signaling pathway [72].
We can draw the conclusion that in mammals H2S could play opposite effects, enhancing or decreasing autophagy induction, which may be attributed to the sulfide concentration, reaction time, cell types and/or differences among the diseases studied. The administration of exogenous H2S in mammalian systems has also typically been performed at micromolar concentrations as in plants [75]. Higher doses of H2S exposed in some publications lead to contradictory data.
However, in all cases, the final outcome of the role of H2S is cell survival, which likewise has been described in plant systems. When stress is mild, in mammals H2S often activates autophagy to protect cells, usually by reducing stress conditions, but with the progression of the disease, H2S can act as a regulator inhibiting autophagy to avoid excess stress-induced autophagy and cell death.
This entry is adapted from the peer-reviewed paper 10.3390/antiox11020327
This entry is offline, you can click here to edit this entry!