Our planet urgently needs sustainable solutions to alleviate the anthropogenic global warming and climate change. Homogeneous catalysis has the potential to play a fundamental role in this process, providing novel, efficient, and at the same time eco-friendly routes for both chemicals and energy production. In particular, pincer-type ligation shows promising properties in terms of long-term stability and selectivity, as well as allowing for mild reaction conditions and low catalyst loading. Indeed, pincer complexes have been applied to a plethora of sustainable chemical processes, such as hydrogen release, CO2 capture and conversion, N2 fixation, and biomass valorization for the synthesis of high-value chemicals and fuels.
1. Introduction
During the last 15 years, organometallic pincer-type complexes have emerged as a highly promising group of catalysts for numerous processes within sustainable chemistry. They have been applied in energy production through hydrogen generation, dehydrogenative synthesis of high-value chemicals, as well as CO
2 and N
2 hydrogenations for carbon dioxide capture and recycling and a more sustainable ammonia production, respectively. As such, the use of this family of homogeneous catalysts enhances the sustainability of an incredible number of chemical processes. High catalytic activity at mild reaction conditions, low catalyst loading, combined with high selectivity and excellent atom efficiency are the general main advantages. Notably, all these aspects are crucial when considering the sustainability of chemical processes, as dictated by the green chemistry guidelines [
1]. Unfortunately, catalyst deactivation and/or degradation are usually the main drawbacks of homogeneous catalysis, otherwise excellent systems in terms of activity, selectivity, and reaction conditions. The currently employed heterogeneous alternatives are more robust and with an established know-how on the processes, but they usually require high temperatures and hence are high-energy demanding. Pincer-type ligations provides increased robustness because of the stabilization of the tridentate coordination, resulting in homogeneous catalytic systems with increased chemical and thermal stability [
2,
3,
4].
The ligand design of pincer complexes offers numerous possibilities as well as potential catalytic applications [
5,
6]. For example, the pincer arm can bear an array of different heteroatoms and functionalities. In addition to affording chemical stability, the pincer ligand can take active part of the catalytic cycle by providing a suitable coordination site for the substrate, weakening selected bonds (H-X bonds), or accepting/donating electrons and protons. Moreover, the cooperation between the central metal atom and the ligand is tunable based on the desired steric/electronic environment and catalytic application.
After the first family of PCP complexes was synthetized by Shaw in 1976 [
7], numerous research groups have applied this concept for almost all types of homogeneously catalyzed chemical reactions. A myriad of novel complexes with different pincer arms have been synthetized and characterized, including a vast family of carbene pincers [
8,
9,
10,
11,
12,
13,
14,
15,
16,
17,
18,
19,
20,
21,
22,
23], PNP [
24,
25,
26,
27,
28,
29], PNN [
30,
31], POP [
32], PCP [
33,
34,
35,
36,
37,
38], SNS [
39,
40], NCN [
41], NSiN [
42], CNC [
43], CNN [
44], NNN [
45], as well as sulfur- [
46,
47,
48], silicon- [
49,
50], selenium- [
51,
52], and boron-functionalized [
53,
54] pincer ligands. Indeed, this topic represents one of the most attractive areas in homogeneous catalysis [
55,
56,
57,
58,
59,
60,
61,
62,
63]. Many of the most promising results in sustainable transformations have been achieved with second and third-row transition metals, such as Ru [
40,
64,
65,
66,
67,
68,
69,
70,
71,
72], Os [
73,
74,
75,
76], Ir [
77,
78,
79,
80,
81], Rh [
82,
83], and Pd [
84,
85,
86,
87,
88,
89]. Nevertheless, the current trend in the scientific community is to identify cheaper alternatives based on earth-abundant metals such as Fe [
90,
91,
92,
93,
94,
95,
96], Mn [
97,
98,
99,
100], Ni [
101,
102,
103,
104,
105], V [
106,
107,
108], and Co [
109,
110,
111,
112,
113,
114]. In particular, iron and manganese PNP pincer complexes show optimal performance in many relevant sustainable transformations in the optic of the hydrogen economy. Several excellent reviews cover this relevant transition toward first-row metals for a more sustainable chemical production [
115,
116,
117,
118,
119,
120,
121,
122,
123,
124].
More recently, the incorporation of pincer complexes into porous materials acting as supports has been investigated using the supported (ionic) liquid phase catalysis (SILP or SLP) [
125,
126]. The idea is to combine the excellent activity of homogeneous systems with the robustness given by the heterogeneous nature of the support. Important examples using pincer-type homogeneous catalysts can be found in aldehyde hydrogenation using Fe(II)-PNP complexes [
127,
128], and continuous-flow alkane dehydrogenation [
129].
Furthermore, several groups have been exploring the use of pincer complexes for a wide series chemical transformations, further expanding the applicability of this family of catalysts. Representative examples include olefination [
130,
131,
132], hydroamination [
133,
134,
135], hydrocarboxylation [
136], hydrovinylation [
137], aminomethylation [
138], dehydrogenation of alkanes [
139,
140,
141,
142,
143,
144], alkane metathetis [
145], N-formylation of amines [
146,
147], C-alkylation of secondary alcohols [
148,
149], α-alkylation of ketones [
150,
151], and alkylation of amines [
152,
153,
154,
155,
156] and anilines [
157].
The deoxydehydration (DODH) of biomass-derived vicinal diols and polyols has also been explored by employing metal pincer complexes. The reaction proceeds in the presence of a sacrificial reducing agent and results in the formation of alkenes, relevant building blocks for the polymer industry. Some examples using pincer ligation are reported with vanadium [
106,
107], rhenium [
158], as well as molybdenum pincer catalysts [
159]. The field is relatively immature and further optimization is necessary. A comprehensive overview of the best catalytic systems for DODH reactions, including the aforementioned pincer complexes, is provided in the detailed reviews of Fristrup [
160] and Monbaliu [
161].
Remarkably, there are several reports in literature using pincer-type metal complexes as suitable catalysts for water splitting reactions. The process is key for the development of the hydrogen economy that requires green and sustainable hydrogen produced via solar or wind energy. Many groups explored various combinations of metals and pincer ligation [
14,
162,
163]; in particular, several works report the use of Milstein Ru-PNP catalyst
3 for this transformation [
164,
165,
166,
167,
168,
169].
2. Dehydrogenation Reactions
2.1. Early Works
The first example of AAD by homogeneous catalysis dates back to 1960s with the work by Charman, using rhodium chloride as catalyst [
198]. In the mid-1970s, Robinson described the ruthenium complex [Ru(OCOCF
3)
2(CO)(PPh
3)
2] in combination with trifluoroacetic acid for the dehydrogenation of isopropanol, 1-butanol, ethanol, methanol, and glycerol [
199,
200,
201,
202]. Several improvements were achieved in the subsequent 20 years, using various type of homogeneous systems in combinations with a range of additives, including light irradiation.
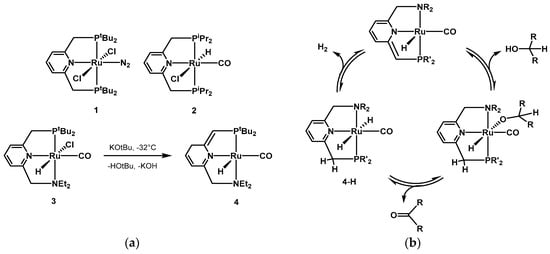
In 2004, Milstein presented the first example of metal–ligand cooperating pincer ligands in AAD for synthetic purposes [
203,
204]. In a series of ruthenium(II)-based complexes, the PNP pincer bearing a pyridine moiety and various phosphine substituted side arms was found to be very active for the dehydrogenation of simple secondary alcohols. Some examples of the first generations of Milstein’s catalysts can be found in
Scheme 1a. In most cases, there is a direct participation of the pincer ligands in the catalytic cycle. The pyridine moiety rearranges by aromatization-dearomatization [
64,
177,
205,
206,
207,
208,
209], facilitating the coordination of the alcoholic substrate and the subsequent hydrogen release from the dihydride species, as depicted in the catalytic cycle in
Scheme 1b. The involvement of the pincer moiety in the catalytic cycle has been investigated by many groups [
210,
211,
212,
213,
214,
215]. In 2015, Li reported computational mechanistic studies on several reactions using the Milstein PNP and PNN catalysts [
216]. The authors recalculated rate-determining steps and investigated the aromatization-dearomatization equilibria. It was found that aromatic PNP and PNN ligands often provide the lowest activation energy for some steps, whereas for other steps, the aromatization–dearomatization process was not involved in the lowest energy pathway. Very recently, Gusev investigated the mechanism of AAD of alcohols, as well as ester hydrogenation, using catalyst
4, and identified the dihydrido complex
4-H (
Scheme 1b) as the active species for both dehydrogenation and hydrogenation reactions [
217].
Scheme 1. (
a) Examples of Milstein’s first generation pincer catalysts [
204]; (
b) example of acceptorless alcohol dehydrogenation (AAD) reaction mechanism using Milstein’s type PNN pincer complexes based on aromatization/dearomatization of the pyridine moiety.
2.2.1. Methanol Dehydrogenation
The homogeneous dehydrogenation of methanol to H
2 and CO
2 in the optic of a methanol-based economy has been intensively studied over the past decade. The topic has been reviewed by Alberico and Nielsen in 2015 [
320], by Prakash in 2018 [
321], and very recently by Araya, Liso, Cui, and Knudsen Kær [
322]. Importantly, methanol is currently produced from fossil fuels through syngas, hence a sustainable production from biomass or/and atmospheric CO
2 is highly desirable (see
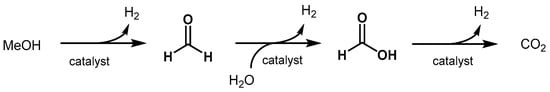
Section 3.1.2). The aqueous reforming of methanol involves three consecutive steps yielding three molecule of hydrogen for each molecule of methanol (
Scheme 9). The first step is the dehydrogenation of methanol to afford formaldehyde and the first equivalent of hydrogen. Then, formaldehyde reacts with water to form methanediol that can undergo a second dehydrogenation resulting in formic acid. The latter is further dehydrogenated to finally produce CO
2 and the third molecule of hydrogen.
Scheme 9. Reaction pathway of aqueous methanol reforming.
The reforming of methanol to produce hydrogen is currently carried out at elevated temperatures (>200 °C) and pressures (>25 bar) by means of heterogeneous catalysts such as Pt/ Al
2O
3, as well as the less expensive Cu/ZnO/Al
2O
3 [
323,
324,
325,
326,
327,
328]. As discussed below, pincer complexes allow the direct release of hydrogen gas from aqueous methanol at temperatures below 100 °C, with low catalyst loadings as well as promising stability properties.
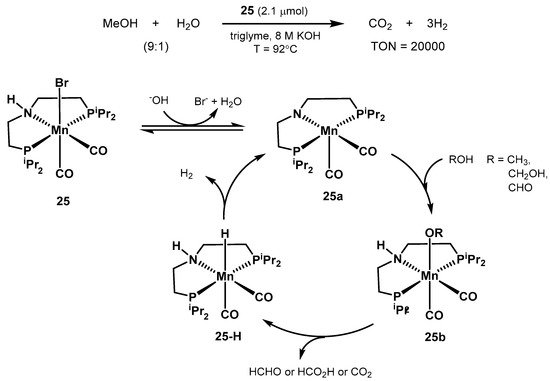
In 2017, Beller proposed the structurally defined manganese complex
25 as an active catalyst for aqueous methanol dehydrogenation (
Scheme 16) [
346]. The optimized conditions resulted in 20,000 turnovers after 900 h at 92 °C, starting from a 9:1 CH
3OH/H
2O mixture in triglyme, with 8M KOH, and in the presence of 10 equivalents of the PNP ligand to the catalyst (2.1 μmol). Moreover, other organic carriers such as ethanol, paraformaldehyde, and formic acid were successfully dehydrogenated as well. Unlike the previous work by Bernskoetter, Hazari, and Holthausen [
336], the presence of Lewis acid additives resulted in no observable improved catalytic activity.
Scheme 16. Manganese-catalyzed methanol reforming and proposed catalytic cycle proposed by the group of Beller [
346].
The same group showed that, similar to the PNP complexes of ruthenium, iron, and manganese, also the iridium-PNP catalyst
8 is able to promote methanol dehydrogenation under mild conditions albeit with lower catalytic activity [
347]. Complex
8 afforded a TON of 1900 after 60 h at 92 °C, showing promising stability over time. In this case too, highly basic conditions were required (8M KOH) for consistent catalytic activity starting from a 9:1 mixture of CH
3OH/H
2O.
In 2019, Beller improved the performance of Ru-PNP catalysts for methanol dehydrogenation using another bi-catalytic system formed by the catalysts
10/
10-Me (
Scheme 13b) [
335]. Based on observations on the formic acid dehydrogenation step [
348], the addition of catalyst
10-Me promotes the rate of hydrogen release from formic acid in the last step. The combination of the two catalysts together was 1.5 times more active than individually. Employing 8.56–9.62 μmol of
10 +
10-Me in a 9:1 MeOH:H
2O mixture in triglyme, with 40 mmol KOH at 92.5 °C, afforded a clean 3:1 H
2/CO
2 mixture with TOF = 1063 h
−1 and a TON = 3189 (calculated based on total amount of catalysts present) after 3 h. This work further shows that applying bi-catalytic systems in cascade reactions is a promising solution to improve the catalytic performance, taking advantage of synergetic effects between two active catalytic species.
2.2.2. Formic Acid Dehydrogenation
The use of formic acid as hydrogen carrier and storage system has been widely investigated and reviewed in various works [
299,
300,
350,
351,
352,
353,
354,
355,
356,
357,
358,
359]. A variety of both heterogeneous and homogeneous catalytic systems have been applied for its decomposition, including the use of light irradiation [
360,
361,
362,
363,
364,
365,
366,
367,
368,
369,
370,
371,
372,
373,
374,
375,
376,
377,
378,
379,
380,
381].
In 2011, following previously reported works on iron-catalyzed formic acid dehydrogenation [
382,
383]. Beller reported the remarkable activity of an iron complex bearing the tetradentate tripodal ligand tris[(2-diphenylphosphino)ethyl]phosphine [P(CH
2CH
2PPh
2)
3 (PP
3) [
384]. The authors tested both the in situ formation of the active species, as well as various synthesized iron hydride complexes bearing the same PP
3 ligand. The activity of the synthetized catalysts was found to be comparable with that of the in situ formed systems in the presence of 2 equivalents of PP
3 ligand. After optimization, simply applying 5 mmol of Fe(BF
4)
2·6H
2O and 2 equivalents of PP
3 to a solution of formic acid in propylene carbonate, afforded a TOF of 9425 h
−1 and a surprising TON of 92,000 at 80 °C.
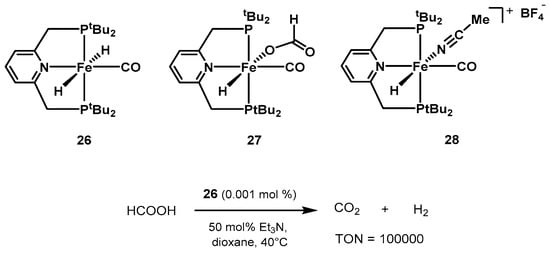
Contemporarily, Milstein was also investigating the performance of iron pincer complexes based on the lutidine moiety, reporting suitable catalysts for the hydrogenation of ketones [
385,
386], as well as CO
2 hydrogenation to formate [
387]. In 2013, the group showed a series of iron pincer complexes as active catalysts for formic acid dehydrogenation (
Scheme 17) [
388]. The dihydrido complex
trans-[Fe-(
tBuPNP)(H)
2(CO)]
26 showed the best performance, reaching TON values up to 100,000 at 40 °C in the presence of trialkylamines. 0.001 mol% of
26 in 1,4-dioxane, in the presence of 50 mol% NEt
3, resulted in full conversion of formic acid after 10 days.
Scheme 17. PNP pincer catalysts screened by Milstein for low-temperature formic acid dehydrogenation [
388].
In 2018, Beller reported that also the known ruthenium dihydride [RuH
2(PPh
3)
4] is a suitable catalyst for the dehydrogenation of aqueous formic acid at low temperature (TOF up to 36,000 h
−1 at 60 °C in THF) [
407]. The catalyst was active for 120 days and it does not require basic additives.
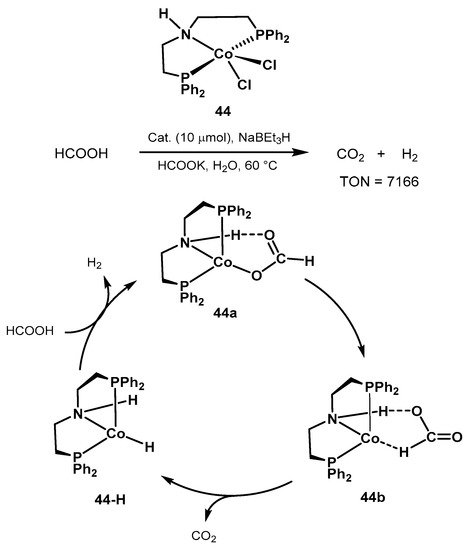
The same group also proposed the cobalt catalyst precursor
44 shown in
Scheme 29 for formic acid dehydrogenation in aqueous media and at mild conditions [
408]. Reactions were performed at 60–80 °C in the presence of HCOOK. Since the catalytically active species are air-sensitive, the authors investigated the in situ activation of the pre-catalyst
44 in the presence of NaBEt
3, resulting in the active hydrido complex
44-H (
Scheme 29). Importantly, under optimized conditions, the benchmark
Ru-MACHO resulted in scarce H
2 evolution, whereas the manganese catalyst
25 showed no activity in aqueous conditions. The authors proposed an outer-sphere mechanistic cycle similar to the classic Ru-PNP catalyst, with the amine proton taking active part in the catalytic cycle. The authors concluded that the rate-determining step is the C-H activation resulting in CO
2 release and formation of the amine complex. The work provides useful information for the development of non-noble metal catalysts for formic acid dehydrogenation.
Scheme 29. Novel cobalt-PNP pincer complex for low-temperature formic acid dehydrogenation proposed by Beller [
408].
3. Hydrogenation Reactions
For case of brevity, the focus will be on processes involving carbon dioxide and dinitrogen as the main substrates of interests, as well as chemical transformations promoting the valorization of biomass-derived molecules. Nevertheless, pincer complexes have achieved remarkable results in the (transfer) hydrogenation of a wide series of substrates such as ketones [
385,
457,
458,
459,
460,
461,
462], esters [
40,
179,
220,
386,
400,
463,
464,
465,
466,
467,
468,
469,
470,
471,
472,
473,
474,
475,
476,
477], aldehydes [
478,
479,
480], amides [
67,
481,
482,
483,
484,
485], and imines [
486,
487].
Carbon dioxide is sadly known to be the main responsible for the anthropogenic climate change and global warming [
488,
489,
490,
491]. At the same time, CO
2 represents an easily accessible C1 building block with the potential to replace the commonly used petrochemical carbon sources in a plethora of useful chemical transformations, dramatically increasing their intrinsic sustainability [
492,
493,
494]. Several approaches have been investigated for its capture from the atmosphere, as well as from localized emission sources [
495,
496,
497,
498,
499,
500,
501,
502,
503,
504]. The desorbed CO
2 is subsequently compressed and stored in underground rock formations or utilized in the direct synthesis of value-added products [
505,
506,
507,
508,
509,
510]. Indeed, the industry already uses several million tons of CO
2 for the production of e.g., urea, salicylic acid, cyclic carbonates, and polypropylenecarbonate [
511,
512,
513].
In the past decades, the catalytic hydrogenation of CO
2 has gained attention as a powerful tool to store green hydrogen, thereby electrical energy, as introduced by the seminal works by Asinger [
514], Leitner [
515,
516], Noyori [
517], as well as Olah [
301,
518,
519,
520]. The process, combined with the aforementioned methanol/formic acid dehydrogenation reactions, has the potential to close the ideal cycle of CO
2-free energy release and storage. Currently, both methanol and formic acid are industrially produced using fossil feedstock via carbon monoxide, which has lower kinetic and thermodynamic stability compared to CO
2. The direct synthesis from CO
2 is traditionally carried out at high temperatures and pressures with heterogeneous metal catalysts such as Cu/ZnO/Al
2O
3 [
521,
522,
523,
524]. Thus, the sustainability of the direct CO
2-route is strictly dependent on the use of green hydrogen produced without contemporary CO
2 release in the atmosphere, as well as catalytic systems operating efficiently under milder reaction conditions. A variety of homogeneous catalytic systems has been employed for the direct hydrogenation of CO
2 into green fuels [
213,
214,
517,
525,
526,
527,
528,
529,
530,
531,
532,
533,
534,
535], including first-row metal complexes [
536,
537,
538,
539,
540,
541,
542,
543,
544]. Pincer-type ligation shows again very encouraging features in terms of stability and mild reaction conditions, with promising possibility of further optimization. In addition, often the same catalyst is active in both directions of hydrogenation and dehydrogenation, expanding the applicability and robustness of this family of catalysts.
3.1. CO2 Hydrogenation
3.1.1. Early Works
The hydrogenation of carbon dioxide by means of homogeneous catalysis has grown extensively in the last decade. An overview of the best performing systems for CO
2 hydrogenation up to 2010 can be found in the work of Beller [
557], while in 2018 and 2019 Prakash reviewed the topic in depth including the use of pincer type complexes [
558,
559]. In this review, we will focus mostly in CO
2 conversion to formic acid (and/or formate salts) as well as to methanol, both of them representing accessible green fuel and hydrogen carriers.
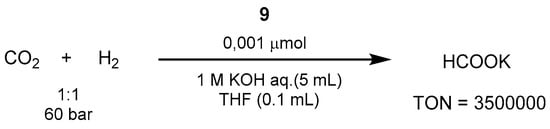
Up to 2010, the best performing catalytic system was represented by the iridium-PNP catalyst
9 (shown in
Scheme 2), reported by Nozaki in 2009, which overcame previously reported Ru [
560,
561,
562,
563], Rh [
564,
565,
566], and Ir [
567] homogeneous systems. In the work by Nozaki, the reactions were carried out in aqueous KOH, resulting in potassium formate (HCOOK) as the product [
568]. Thus, using the trihydridoiridium(III)-PNP complex
6 at 120 °C and 60 bar of 1:1 CO
2/H
2 in 1.0 M aqueous KOH it was possible to achieve excellent TON and TOF values of 3,500,000 and 150,000 h
−1, respectively (
Scheme 37).
Scheme 37. CO
2 hydrogenation with Ir-PNP catalyst reported by Nozaki in 2009 [
568].
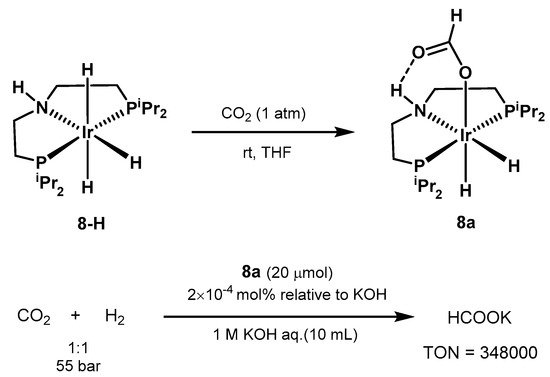
The use of iridium pincer complexes was further explored by Hazari in 2011 [
569]. The authors were able to isolate the air and moisture stable catalyst
8a, obtained from
8-H in the presence of CO
2. Under optimized conditions, the system resulted in yields up to 70% in formate, with TON of 348,000 and TOF of 14,500 h
−1 (
Scheme 38).
Scheme 38. CO
2 hydrogenation with Ir-PNP catalyst proposed by Hazari [
569].
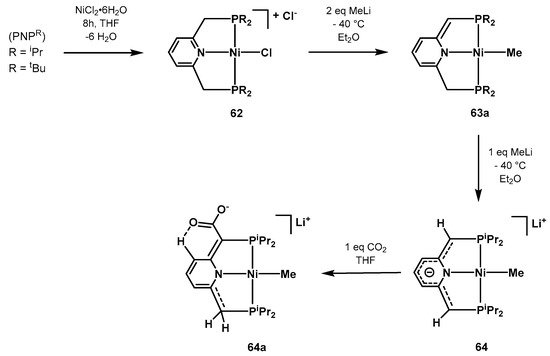
In 2013, Milstein synthetized and characterized a series of novel pyridine-based Ni-PNP complexes with the aim of investigating the aromatization/dearomatization equilibria prompted by double protonation-deprotonation of the pincer arm [
583]. Complex
64 can be readily prepared starting from NiCl
2·6H
2O and the PNP
R ligand in a cascade reaction as depicted in
Scheme 42. Complex
64, where the negative charge is delocalized in the pincer moiety, was characterized by single-crystal X-ray diffraction studies. In the presence of CO
2, it undergoes electrophilic attack resulting in the anionic specie
64a, observed by NMR spectroscopy.
Scheme 42. Synthesis and CO
2 coordination of the Ni-PNP catalyst
64 proposed by Milstein [
583].
Goldman performed DFT studies on the hydrogenation of dimethyl carbonate to methanol using the Milstein-type PNN catalyst
4 [
215]. The work provided new insights on the mechanism for the C-OMe bond cleavage. The authors proposed an ion-pair-mediated metathesis pathway in which the formed alkoxide C–H bond binds to the ruthenium atom. By simple reorientation of the dimethoxymethanoxide anion formed upon transfer of a hydride to dimethyl carbonate, the methoxy group of the [OCH(OMe)
2]
− anion is in a close proximity to the metal, allowing the C–OMe bond cleavage.
3.1.2. CO2 Hydrogenation to Methanol
Himeda and Laurenczy reported in 2016 the iridium complex [(Cp*)Ir(dhbp)(OH
2)][SO
4] (dhbp = 4,4′-dihydroxy-2,2′-bipyridine), already showed for bicarbonate hydrogenation to formate and formic acid dehydrogenation [
584], for the production of methanol from CO
2 at room temperature as well [
585]. The catalyst was active for both CO
2 hydrogenation to formic acid in acidic media, without additives, as well as formic acid disproportionation into methanol, achieving 98% conversion and 96% selectivity after 72 h. Formic acid was obtained by pressurization of a solution of the catalyst with 20 bar CO
2 and 50 bar H
2 at ambient temperature. Methanol was observed by only increasing the temperature, whereas the addition of an optimized sulfuric acid concentration resulted in the enhanced activity of the system. No carbon monoxide was observed, indicating that decarbonylation of formic acid did not occur.
In 2015, Sanford proposed a ruthenium-catalyzed hydrogenation of CO
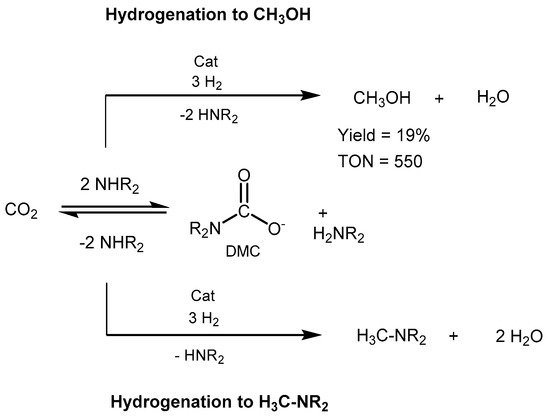
2 to methanol with dimethylamine as capturing agent [
586]. The reduction proceeds in basic conditions through the in situ formation of dimethylammonium dimethylcarbamate (DMC) as a key intermediate (
Scheme 43).
Ru-MACHO-BH (0.03 mol%) was used as catalyst, in the presence of 2.5:50 bar of CO
2/H
2, and 0.25 mmol of K
3PO
4. The reaction afforded a TON of 550 in methanol and an overall 82% conversion of CO
2 to methanol and a mixture of dimethylformamide and dimethylammonium formate, which raises to 96% total conversion when applying 0.1 mol% of catalyst loading. The work represented a step forward toward the integrated CO
2 capture and direct conversion to methanol.
Scheme 43. CO
2 capture and conversion to MeOH proposed by Sanford [
586].
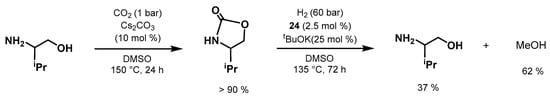
The same year, Milstein developed an innovative methodology for indirect CO
2 hydrogenation by means of prior capture by amino alcohols at low pressure followed by the hydrogenation of the generated oxazolidinone to form MeOH (
Scheme 44) [
587]. Moreover, this approach is inspired by the CO
2 capture industry that uses amino alcohols to capture CO
2 from waste streams [
588]. The work provides new possibilities for the use of oxazolidinone, which can be useful for the production of a liquid fuel such as MeOH. Three Ru-NNP complexes (
3,
24, and
54) were tested for this transformation, with complex
24 being the most active catalyst. The authors screened both 2-(methylamino)ethanol and valinol for the first step of CO
2 capture; valinol was chosen considering its ability to capture CO
2 more selectively under only 1 bar of CO
2. Employing Cs
2CO
3 (10 mol%), DMSO as solvent, and at 150 °C under 1 bar of CO
2, for 24 h afforded the oxazolidinone in >90% yield. The product is not purified or isolated and the leftover CO
2 is simply removed under vacuum. Then, the subsequent hydrogenation of the formed oxazolidinone was performed with
24 (2.5 mol%) and KOtBu (25 mol%) under 60 bar of H
2, at 135 °C, for 72 h, producing MeOH and the amino alcohol precursor in 37% and 62% yields, respectively. Remarkably, the process allows direct CO
2 capture with consequent MeOH formation, avoiding the energy-demanding steps of CO
2 regeneration from capture products and subsequent pressurization.
Scheme 44. Milstein’s CO
2 capture/hydrogenation to MeOH using amino alcohol [
587].
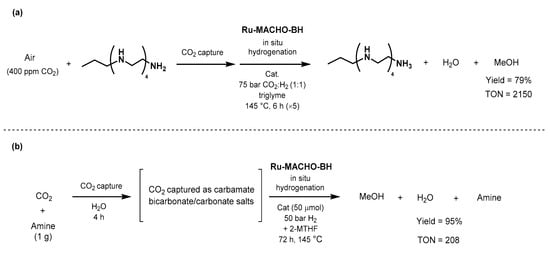
The year after, Olah and Prakash reported an efficient catalytic system for the one-pot CO
2 capture and conversion to methanol, using polyamine and
Ru-MACHO-BH (
Scheme 45a) [
589]. The first step of CO
2 capture was achieved by bubbling synthetic air (400 ppm of CO
2 in N
2/O
2 80/20) in an aqueous solution of pentaethylenehexamine (PEHA) for 64 h.
Ru-MACHO-BH (20 μmol) was applied in the presence of 50 bar of H
2 in triglyme, and 6% of the captured CO
2 (5.4 mmol) was converted into methanol after 55 h in 79% yield (determined by NMR). The authors confirmed the robustness of the method by recycling the catalyst over five consecutive runs of 5 h without significant loss of activity; only 20 μmol of catalyst afforded an overall TON of 2150 at 145 °C under 75 bar pressure of a 1:9 mixture of CO
2:H
2.
Scheme 45. CO
2-capture and direct conversion to methanol using
Ru-MACHO-BH as showed by Olah (
a) and Prakash (
b) [
589,
590]
3.1.3. CO2 Hydrogenation to Formate Salts
Several groups explored the homogeneous hydrogenation of CO
2 to give basic formate salts, which can be later acidified to provide formic acid. Various combination of ruthenium complexes bearing phosphine ligands were found to be active in this transformation [
623,
624,
625]. Important milestones were achieved by Jessop, with Ru-P(Me
3)
3 catalysts [
560,
562], Zhang with Ru-P(Ph
3)
3 species [
626], Leitner with the meta-trisulfonated triphenylphosphine ligand (TPPMS) [
627], as well as Beller, using [RuCl
2(benzene)]
2 in combination with the phosphorous ligand bis(diphenylphosphino)methane (DPPM) [
628]. In addition, the hydrogenation of bicarbonates (used as CO
2-trapping agents) as an indirect route to provide formates has also been investigated by many groups [
629,
630,
631]. Important advances in this regard have been achieved by Beller and Laurenczy using again [RuCl
2(benzene)]
2 and DPPM [
632,
633], resulting in a formate yield of 61%, and a TON = 2,793 after 20 h at 100 °C in the presence of 50 bar of hydrogen and 35 bar of CO
2 in a sodium bicarbonate solution.
In 2012, the same group showed that not only ruthenium, but also iron and cobalt catalysts are highly active for the hydrogenation of bicarbonates to give formates. In the presence of an iron(II)-fluoro-tris(2-(diphenylphosphino)phenyl)phosphino]tetrafluoroborate complex, obtained in situ from Fe(BF
4)
2·6H
2O and tris(2-(diarylphosphino)aryl)phosphine, the hydrogenation of bicarbonates afforded TON > 7500 and TOF > 750, with 77% yield of sodium formate under 60 bar of hydrogen in methanol at 100 °C [
634]. The hydrogenation of CO
2 to formic acid in the presence of methanol and amines was also investigated, resulting in a mixture of formic acid and methyl formate, albeit with only 15% of carbon dioxide conversion.
Using the same approach, the hydrogenation of bicarbonate and carbon dioxide was also demonstrated using an in situ formed cobalt complex [
635]. By simply applying 0.028 mol of Co(BF
4)
2·6H
2O, in the presence of 1 equivalent of the Tetraphos ligand PP
3 (P(CH
2CH
2PPh
2)
3), under 1:1 60 bar of H
2/CO
2 at 80 °C, it was possible to obtain 94% yield of sodium formate, with a TON of 645 after 20 h.
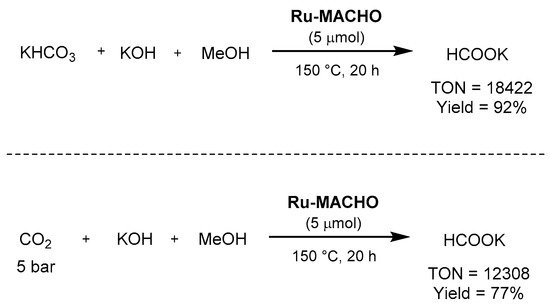
Regarding the use of pincer complexes, Beller employed in 2014
Ru-MACHO as the catalyst for the combined methanol dehydrogenation and bicarbonate hydrogenation, as well as for the synthesis of potassium formate from carbon dioxide, potassium hydroxide, and methanol [
636]. In the first transformation, the reaction afforded excellent TON (>18,000), TOF (>1300 h
−1), as well as yield (92% of potassium formate), as shown in
Scheme 62. Under the same reaction conditions, with 5 bar of carbon dioxide, the reaction afforded a formate yield of 77%, with a TON of 12,308.
Scheme 62. Ru-MACHO-catalyzed hydrogenation of bicarbonate and CO
2 to formate showed by Beller [
636].
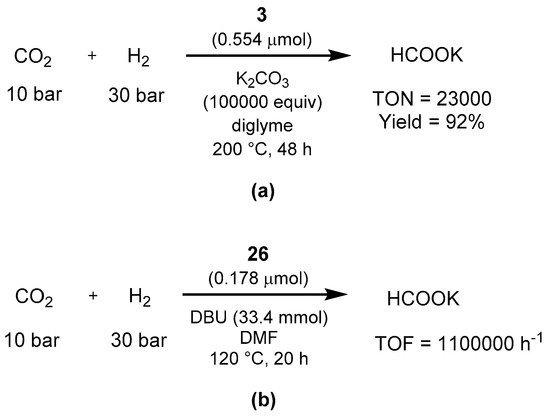
Sanford showed in 2013 that the Milstein PNN catalyst
3 efficiently catalyze the hydrogenation of CO
2 to formate in the presence of potassium carbonate as base (
Scheme 63a) [
637]. In addition, the authors performed stoichiometric studies of various organometallic intermediates, showing that the reaction proceeds with a classic ligand-based aromatization/dearomatization pathway. The year after, Pidko showed that another Milstein catalyst, the PNP-pincer
26, is able to perform the same transformation [
638]. With the reaction conditions shown in
Scheme 63b, the hydrogenation of CO
2 proceeds in the presence of DBU as base, showing activity already at 65 °C and a TOF of 1,100,000 h
−1 at 120 °C (
Scheme 63b). Importantly for an energetic cycle point of view, catalyst
26 is very active in the reverse formic acid dehydrogenation as well, affording high turnover numbers (1063000) in DMF/NEt
3.
Scheme 63. Hydrogenation of CO
2 to formate using Milstein’s catalysts
3 (
a) and
26 (
b) as reported by Sanford [
637] and Pidko [
638].
This entry is adapted from the peer-reviewed paper 10.3390/catal10070773