Several randomized, double blind, placebo-controlled trials (RCTs) have demonstrated that low-density lipoprotein cholesterol (LDL-C) lowering by using statins, including high-doses of strong statins, reduced the development of cardiovascular disease (CVD). However, among the eight RCTs which investigated the effect of statins vs. placebos on the development of CVD, 56–79% of patients had the residual CVD risk after the trials. In three RCTs which investigated the effect of a high dose vs. a usual dose of statins on the development of CVD, 78–87% of patients in the high-dose statin arms still had the CVD residual risk after the trials. An analysis of the characteristics of patients in the RCTs suggests that elevated triglyceride (TG) and reduced high-density lipoprotein cholesterol (HDL-C), the existence of obesity/insulin resistance, and diabetes may be important metabolic factors which determine the statin residual CVD risk.
1. Introduction
A great number of epidemiological studies, including the Framingham study, have shown that an elevation of serum low-density lipoprotein cholesterol (LDL-C) increases the incidence and death from cardiovascular disease (CVD), such as coronary heart disease (CHD). The 4S, a randomized trial of cholesterol lowering in 4444 patients with CHD, reported in 1994, showed that the decrease in LDL-C by using simvastatin made a significant reduction of CVD by 35% [
1]. The WOSCOPS, which studied the primary prevention of CVD by statin, showed that pravastatin lowered LDL-C levels by 26%, and reduced coronary events (nonfatal myocardial infarction (MI) or death from CHD) by 31% in the pravastatin group [
2]. After the publication of the WOSCOPS, the ASCOT-LLA [
3], AFCAPS/TexCAPS [
4], CARDS [
5], and JUPITER [
6] studies showed a primary prevention of CVD by statins. In addition to the 4S, the LIPID [
7] and CARE [
8] studies demonstrated a secondary prevention of CVD by statins.
In the 2000s, strong statins showing an intense lowering effect of LDL-C appeared, and randomized, double blind, placebo-controlled trials (RCTs) to study the effect of high doses and usual doses of statins on the development of cardiovascular events have been performed. The PROVE IT-TIMI 22 study allocated patients with acute coronary syndrome (ACS) to intensive (daily 80 mg of atorvastatin) and moderate (daily 40 mg of pravastatin) lipid-lowering therapy [
9]. An intensive lipid-lowering therapy using atorvastatin decreased LDL-C by 51% and reduced cardiovascular events by 16%. This study demonstrated that, in patients who have recently had ACS, an intensive lipid-lowering statin therapy induced a greater protection against death or major cardiovascular events as compared with a standard therapy, indicating that ACS patients obtain a benefit from the early and continuous lowering of LDL-C to levels substantially below the previous target level. In the IDEAL study, patients with a previous MI were randomly assigned to receive a high dose of atorvastatin (80 mg/day) or a usual dose of simvastatin (20 mg/day) [
10]. The mean LDL-C levels were 104 mg/dL in the simvastatin group and 81 mg/dL in the atorvastatin group. Major cardiovascular events occurred in 608 patients in the simvastatin group and 533 patients in in the atorvastatin group (hazard ratio (HR), 0.87; 95% confidence interval (CI); 0.77–0.98;
p = 0.02). In the TNT study, the efficacy and safety of lowering the LDL-C levels below 100 mg/dL were evaluated in patients with CHD [
11]. Patients were randomly assigned to a double-blind therapy and received either daily 10 mg or 80 mg of atorvastatin. The LDL-C levels were 77 mg/dL during treatment with 80 mg atorvastatin and 101 mg/dL during treatment with 10 mg atorvastatin. The HR for an absolute reduction in the rate of major cardiovascular events was 0.78, proposing an intensive lipid-lowering therapy with daily 80 mg of atorvastatin in patients with CHD provides a significant clinical benefit beyond that afforded by the treatment with daily 10 mg of atorvastatin.
The Cholesterol Treatment Trialists’ (CTT) Collaboration performed a prospective meta-analysis of data from 90,056 individuals in 14 RCTs of statins [
12]. There was a 12% proportional reduction in all-cause mortality and a 21% proportional reduction in major vascular event per mmol/L reduction in LDL-C. The proportional reduction in major vascular events differed significantly according to the absolute reduction in LDL-C achieved. These results emphasized the need to consider prolonged statin treatment with substantial LDL-C reduction in patients at high CVD risk. Furthermore, the CTT undertook meta-analyses of individual participant data from RCTs involving more versus less intensive statin regimens (five trials) and of statin versus control (twenty-one trials) [
13]. When statins deceased LDL-C by 1.0 mmol/L (38.7 mg/dL), reductions in major vascular events were obtained by 22%. Further, all-cause mortality was reduced by 10% by a 1.0 mmol/L LDL reduction, largely reflecting the significant reductions in deaths due to CHD and other cardiac causes. For LDL-C, “the lower, the better” was suggested to be correct to reduce the risk of cardiovascular events.
Therefore, the current treatment guidelines focus on LDL-C by using statins for reducing CVD risk [
14,
15,
16,
17,
18]. However, the PROVE IT-TIMI 22 study and the TNT study showed that CVD events occurred in a substantial number of patients, despite having LDL-C levels below 80 mg/dL [
9,
11]. These recent studies have shown that the risk of development of CVD remains even when statins are used to strongly reduce LDL-C, and such a risk has become to be called the statin residual CVD risk.
2. Properties and Normal Metabolism of Lipoproteins
To understand the association between lipid abnormalities and the development of atherosclerosis, which induces CVD, first we show the profile of the lipoproteins and their normal metabolism.
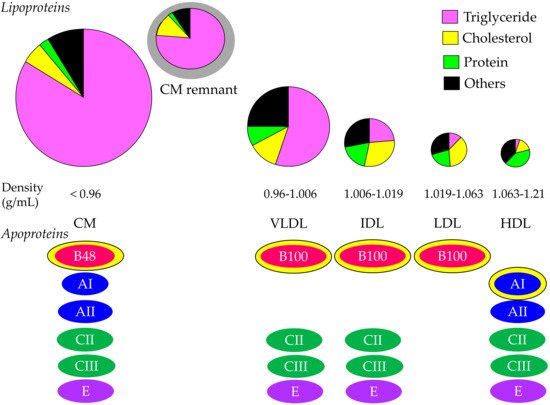
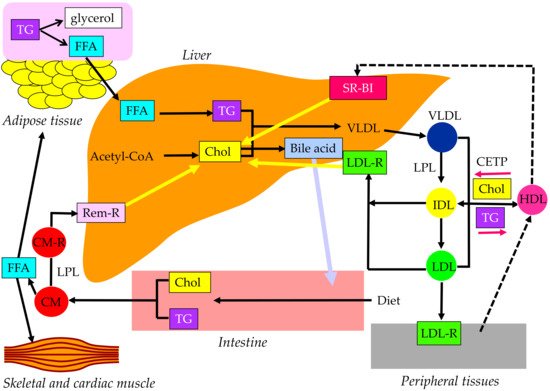
Cholesterol and TG are insoluble in water; therefore, these lipids must be transported in association with apoproteins. Lipoproteins are complex particles with a central core containing cholesterol esters (CE) and TG surrounded by free cholesterol (FC), phospholipids, and apoproteins. The compositions and apoproteins included in each lipoprotein are shown in Figure 1. The metabolism of the lipoproteins is shown in Figure 2.
Figure 1. Composition and apoproteins included in each lipoprotein. Yellow–bordered apoproteins indicate that they are the main apoproteins in each lipoprotein. CM, chylomicron; HDL, high–density lipoprotein; IDL, intermediate–density lipoprotein; LDL, low–density lipoprotein; VLDL, very–low–density lipoprotein.
Figure 2. Metabolism of lipoproteins. CETP, cholesterol ester transfer protein; CM, chylomicron; CM-R, chylomicron receptor; Chol, cholesterol; FFA, free fatty acid; HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LDL-R, low-density lipoprotein receptor; LPL, lipoprotein lipase; Rem-R, remnant receptor; SR-BI, scavenge receptor BI; TG, triglyceride; VLDL, very-low-density lipoprotein.
Chylomicron (CM) is the largest and lowest density lipoprotein synthesized and secreted from the small intestine after the ingestion of dietary fat. More than 80% of the composition of CM is TG. The main apoprotein of CM is apo B48, which is synthesized by the small intestine. After the release from the intestine into the lymphatic system, newly secreted CM enter the systemic circulation via the thoracic duct. CM obtains apo C-II and apo C-III from HDL in plasma. TG in CM is hydrolyzed by lipoprotein lipase (LPL), which requires apo C-II to have a normal function. TG is hydrolyzed to free fatty acid (FFA), which is used in skeletal and cardiac muscles as an energy source and/or is stored in adipose tissue in the form of TG. Reduction of TG in CM by LPL makes the CM remnant, which is taken up by the remnant receptor via apo E in the liver.
VLDL has apo B100, which is synthesized by the liver. Approximately 55% of VLDL is TG. Once released from the liver, TG is carried in VLDL and is metabolized in the muscle and adipose tissue by LPL, releasing FFA and resulting in IDL formation. IDL are metabolized to LDL, which are taken up by the LDL receptor in tissues, including the liver. The proportion of TG in IDL and LDL are 24% and 12%, respectively, and the proportion of cholesterol in IDL and LDL are 29% and 37%, respectively. CM, CM remnant, VLDL, and IDL are classified as TG-rich lipoproteins. Approximately 25–75% of VLDL remnants are not uptaken by the liver, but rather are rather changed to LDL [
19].
CM is an extrinsic lipoprotein, whereas VLDL, IDL, and LDL are endogenous lipoproteins. VLDL, IDL, and LDL have apo B-100; therefore, such lipoproteins are called apo B-100-containing lipoproteins.
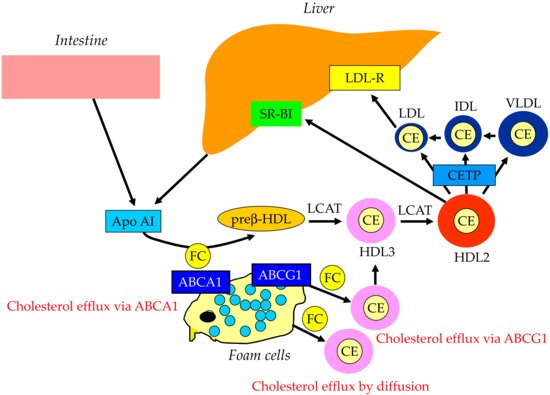
The reverse cholesterol transport by HDL is shown in Figure 3. Apo AI, the main apoprotein of HDL is produced in the liver and the small intestine and receives FC via the ATP-binding cassette transporter A1 (ABCA1), present on the surface of the cell membranes of foam cells, and becomes primitive HDL, preβ-HDL. This process is called cholesterol efflux. ABCG1, another type of ABC transporter, is involved in cholesterol efflux and mediated by HDL2 and HDL3, which are relatively lipid-rich, rather than lipid-free apo AI, unlike ABCA1. In addition to the ABC transporter-mediated pathway, the lecithin–cholesterol acyltransferase (LCAT) lowers the FC concentration on the HDL surface and forms a concentration gradient, which is cholesterol efflux by passive diffusion. As the reverse cholesterol transfer mechanism after cholesterol efflux, there is a pathway mediated by SR-BI and a pathway mediated by apo B-containing lipoproteins. In the former pathway, mature HDL is taken up by the liver via SR-BI. SR-BI binds HDL, but HDL itself is not incorporated and only CE contained in HDL is selectively incorporated into the liver. In the latter pathway, CE in HDL is transferred by CETP to VLDL, IDL, and LDL, which is then taken up by the liver via the LDL receptor and metabolized to bile acids.
Figure 3. Reverse cholesterol transport. ABCA1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; CE, cholesterol ester; CETP, cholesterol ester transfer protein; FC, free cholesterol; HDL, high-density lipoprotein; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LDL-R, low-density lipoprotein receptor; LCAT, lecithin–cholesterol acyltransferase; SR-BI, scavenge receptor BI; VLDL, very-low-density lipoprotein.
3. What Is the Statin Residual CVD Risk?
In addition to elevated LDL-C, hypertriglyceridemia and reduced HDL-C are also dyslipidemia involved in the development of arteriosclerosis. In Japan, hypertriglyceridemia and reduced HDL-C were defined as fasting TG > 150 mg/dL and HDL-C < 40 mg/dL, respectively. Such dyslipidemia was commonly observed in the metabolic syndrome; therefore, it was included in the diagnostic criteria for the metabolic syndrome in Japan.
The meta-analysis to investigate the association between the treatment-induced change in HDL-C and total death, CHD death, and the events adjusted for changes in the LDL-C and drug classes in the RCTs of lipid-modifying interventions was performed [
20]. Available data suggest that simply increasing the amount of circulating HDL-C does not reduce the risk of CHD events, CHD deaths, or total deaths. The presence of accompanying hypertriglyceridemia and insulin resistance may be more important for the development of CVD than the HDL value.
In the EMPATHY study, 5042 patients with hypercholesterolemia with diabetic retinopathy, who had no history of CHD, were allocated to the intensive lipid-lowering group (LDL-C < 70 mg/dL) and the usual lipid-lowering group (100 mg/dL ≤ LDL-C < 120 mg/dL) [
21]. In the subanalysis of the EMPATHY study, the HR of the fourth quartile of TG (135–185 mg/dL) for the development of CVD was 1.65 (95% CI, 1.02–2.65;
p = 0.0331) [
22], suggesting that the serum TG level of 135 can be the residual CVD risk. The study examined the relationship of fasting TG levels to outcomes after ACS in patients treated with statins. Long-term and short-term relationships of TG to risk after ACS were examined in the dal-OUTCOMES trial and the atorvastatin arm of the MIRACL (Myocardial Ischemia Reduction with Acute Cholesterol Lowering) trial, respectively [
23]. Among the patients with ACS treated effectively with statins, fasting TG predicted long-term and short-term cardiovascular risk. Interestingly, the risk for the development of the recurrence of CVD during a short period after ACS was significantly increased in the quartile with the TG of 135–195 mg/dL as compared with the lowest quartile (MIRACLE), and the risk during a long period after ACS was significantly increased in the quartile with the TG of 103–130 mg/dL as compared with the lowest quartile (dal-OCOMES) [
23], indicating that the serum TG level of 103 can be the residual CVD risk.
To understand the statin residual metabolic CVD risk, we evaluated body mass index (BMI) at baseline and serum TG and HDL-C levels achieved after the trials in eight RCTs which investigated the effect of statins on the development of CVD (
Table 1) and three RCTs which investigated the effect of the high dose and usual dose of statins on the development of CVD (
Table 2). A BMI > 25 kg/m
2 and the prevalence of diabetes > 10% at baseline, and HDL-C < 40 mg/dL after the trials, were considered to be the residual metabolic CVD risk. According to the result of the dal-OCOMES [
23], a serum TG > 103 mg/dL was considered to be the residual metabolic CVD risk.
Table 1. Randomized, double blind, placebo-controlled trials to study the effect of statins on development of caridovascular events.
| |
Primary Prevention |
Secondary Prevention |
| Trials |
WOSCOPS [2] |
ASCOT-LLA [3] |
AFCAPS/
TexCAPS [4] |
CARDS [5] |
JUPITER [6] |
4S [1] |
LIPID [7] |
CARE [8] |
| Used statins and daily dose |
pravastatin 40 mg |
atorvastatin 10 mg |
lovastatin 20–40 mg |
atorvastatin 10 mg |
rosuvastatin 20 mg |
simvastatin 20 mg |
pravastatin 40 mg |
pravastatin 40 mg |
| Follow-up (years) |
4.9 |
3.3 |
5.2 |
4 |
1.9 |
5.4 |
6.1 |
5 |
| Risk reduction of cardiovascular events (statins vs. placebo, %) |
31 |
21 |
37 |
37 |
44 |
35 |
29 |
24 |
| Risk reduction of cardiovascular events/year (%) |
6.3 |
6.4 |
7.1 |
9.3 |
23.2 |
6.5 |
4.8 |
4.8 |
| Residual CVD risk (%) |
69 |
79 |
63 |
63 |
56 |
65 |
71 |
76 |
| Residual CVD risk/year (%) |
93.7 |
93.6 |
92.9 |
90.8 |
76.8 |
93.5 |
95.2 |
95.2 |
| Achieved serum lipid levels |
|
|
|
|
|
|
|
|
| LDL-C (mg/dL) |
142 |
90 |
115 |
81 |
55 |
122 |
113 |
97 |
| HDL-C (mg/dL) |
46 |
51 |
39 |
49 |
50 |
49 |
38 |
NA |
| TG (mg/dL) |
142 |
114 |
143 |
143 |
99 |
119 |
126 |
NA |
| Baseline data |
|
|
|
|
|
|
|
|
| BMI (kg/m2) |
26 |
28.6 |
27.1 (men)
26.4 (women) |
28.7 |
28.4 |
26 |
BMI > 30kg/m2, 18% |
28 |
| Prevalence of diabetes (%) |
1 |
24.3 |
6.8 |
100 |
0 |
5 |
9 |
14 |
Table 2. R+A1:D11andomized, double blind, placebo-controlled trials to study the effect of high-dose and usual-dose of statins on the development of cardiovascular events.
| Trials |
PROVE-IT TIMI22 [9] |
IDEAL [10] |
TNT [11] |
| Used statins and daily dose |
pravastatin 40 mg vs. atorvastatin 80 mg |
simvastatin 20 mg vs. atorvastatin 80 mg |
atorvastatin 10 mg vs. atorvastatin 80 mg |
| Follow-up (years) |
2 |
4.8 |
4.9 |
| Risk reduction of cardiovascular events (high-dose vs. usual-dose, %) |
16 |
13 |
22 |
| Risk reduction of cardiovascular events/year (%) |
8 |
2.7 |
4.5 |
| Residual CVD risk (%) |
84 |
87 |
78 |
| Residual CVD risk/year (%) |
92 |
97.3 |
95.5 |
| Achieved serum lipid levels |
|
|
|
| LDL-C |
95 vs. 62 |
99.8 vs. 80 |
101 vs. 77 |
| HDL-C |
42 vs. 40 |
50.6 vs. 50.1 |
apprpximately 48 vs. 49 |
| TG |
175.8 vs. 136.2 |
137.2 vs. 118.5 |
apprpximately 140 vs. 160 |
| Baseline data |
|
|
|
| BMI (kg/m2) |
29.6 vs. 29.6 |
27.3 vs. 27.3 |
28.6 vs. 28.4 |
| Prevalence of diabetes (%) |
18 vs. 18 |
12 vs. 12 |
15 vs. 15 |
Among the eight RCTs which investigated the effect of statins on the development of CVD, 56–79% of patients had the residual CVD risk after the trials. In these studies, the annual residual CVD risk was surprisingly high, with 76.8–95.2%. In the three RCTs which investigated the effect of the high dose vs. usual dose of statins on the development of CVD, 78–87% of patients in the high-dose statin arms still had the CVD residual risk after the trials. In these studies, the annual residual CVD risk was extremely high, with 92–97.3%.
Serum HDL-C < 40 mg/dL and TG > 103 mg/dL after the trials were observed in approximately 18% and 82% of RCTs, and a BMI > 25 kg/m
2 at baseline was observed in all RCTs. The prevalence of diabetes was over 10% in three of eight RCTs which investigated the effect of statins on the development of CVD (37.5%), and the prevalence of diabetes was over 10% in all RCTs which investigated the effect of the high dose vs. usual dose of statins on the development of CVD. Atherogenic dyslipidemia, such as elevated TG and reduced HDL-C, diabetes, and insulin resistance, which are induced by obesity, may be important metabolic factors which determine the statin residual CVD risk [
24,
25].
4. Current Views on Estimation of Residual Cardiovascular Risk in Patients Treated with Statins
In the guidelines on the management of CVD, LDL-C remains the primary target while apo B and non-HDL-C can be secondary targets. Atherogenic TG-rich lipoproteins such VLDL and IDL, and LDL, have apo B-100. The serum level of non-HDL-C is calculated by subtracting HDL-C from total cholesterol; therefore, non-HDL also includes atherogenic TG-rich lipoproteins (VLDL, IDL and remnant) and LDL. Johannesen CDL et al. studied to determine if elevated apo B and/or non-HDL-C are superior to elevated LDL-C in identifying statin-treated patients at residual risk of all-cause mortality and MI. In total, 13,015 statin-treated patients from the Copenhagen General Population Study were included, with 8 years median follow-up. High levels of apo B and non-HDL-C were associated with an increased risk of all-cause mortality and MI, whereas no such associations were found for high LDL-C, suggesting that elevated apo B and non-HDL-C, but not LDL-C, are associated with residual risk of all-cause mortality and MI in statin-treated patients [
63].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23073418