The plasma kinetics involves elementary processes by which free electrons ultimately activate weakly reactive molecules, such as carbon dioxide or methane, thereby potentially starting prebiotic reaction chains. These processes include electron–molecule reactions and energy exchanges between molecules. They are basic processes, for example, in the famous Miller-Urey experiment, and become relevant in any prebiotic scenario where the primordial atmosphere is significantly ionized by electrical activity, photoionization or meteor phenomena. The kinetics of plasma displays remarkable complexity due to the non-equilibrium features of the energy distributions involved.
- Miller-Urey experiment
- prebiotic chemistry
- plasma kinetics
- electrons prebiotic molecules
1. Introduction
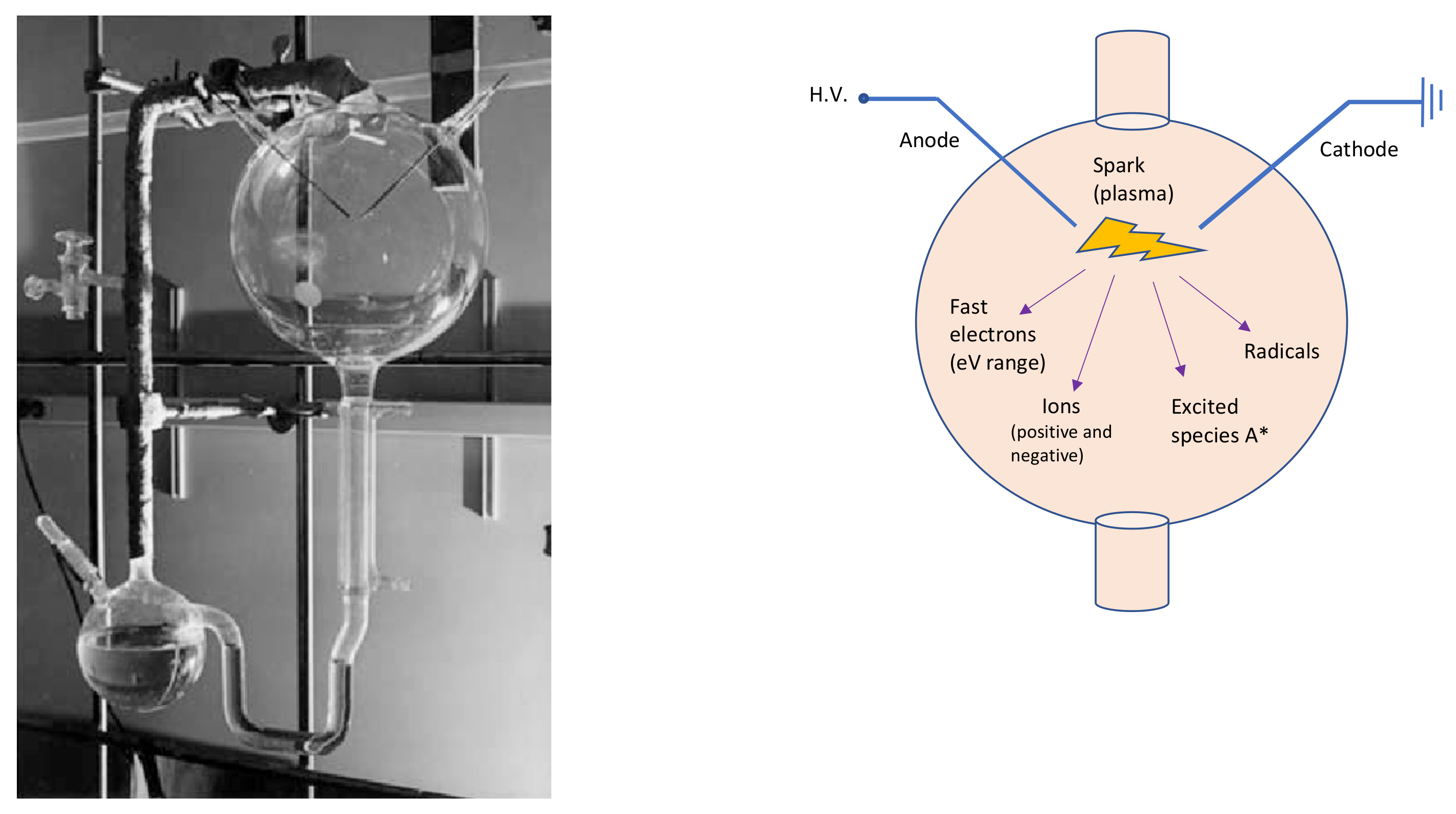
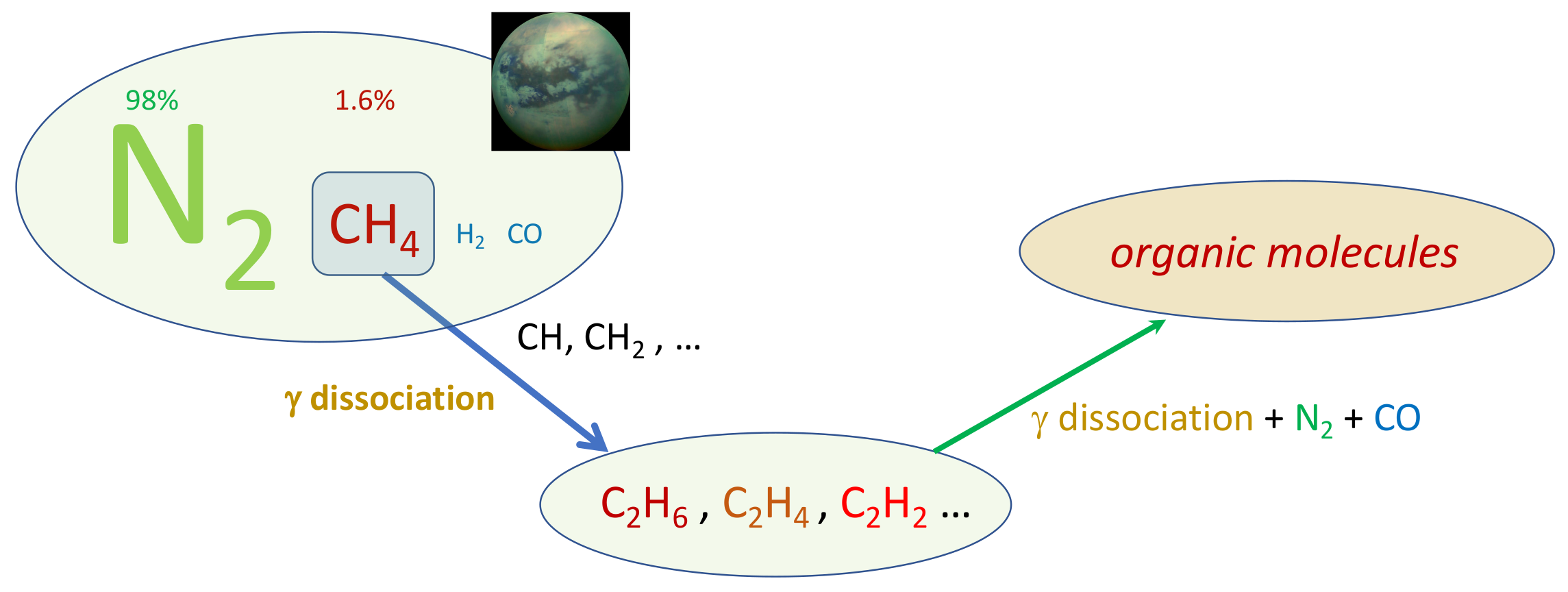
2. Tools from Plasma Kinetics
3. Kinetics of Molecular Activation by Plasma Electrons
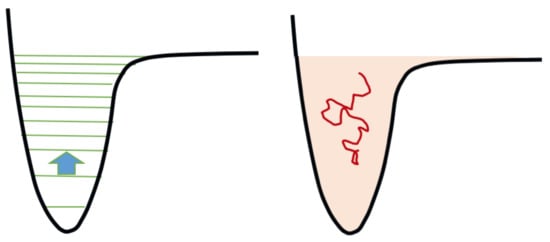

-
A highly reduced mixture of gases including CH4, NH3 and H2O, with molar fractions of 0.7, 0.2 and 0.1, respectively. This composition is taken as representative of Miller-Urey-type experiments and it has also been considered in recent experimental investigation by Scherer and co-authors [85].
-
A mildly reduced mixture of gases dominated by N2 and CO2, with traces of CO, H2O and H2. The molar fractions of the gases are 0.7 for N2, 0.2 for CO2, 0.05 for H2O, 0.025 for CO and for H2. This composition is representative of a prebiotic Earth atmosphere (around 3.8 Gy) at sea level, as suggested in the pioneering study by Kasting [35]. At higher altitudes, traces of O2 and O are also formed from photo-dissociation of CO2 [35].
-
Methane: Cross sections for CH4 are taken from the Magboltz code version 11.9 developed by Biagi [87].
-
Ammonia: Cross sections for NH3 are taken from the Hayashi database of the LXCat database [88].
-
Water vapour: Cross sections for H2O are taken from Biagi’s code Magboltz version 11.9 [87].
-
Carbon dioxide: Electron impact cross sections for CO2 are taken from the Biagi database of LXCat [89]. The elastic momentum transfer cross sections from the Biagi database is corrected to take into account the population of vibrational bending mode levels and quadrupole rotational collisions, according to Vialetto and co-authors [83].
-
Carbon monoxide: Electron impact cross sections for CO are the ones from the Magboltz source code v 11.9 [87]. In [90], this set has been used for calculations of electron transport parameters in CO, with particular focus on the treatment of dipole rotational collisions. A Morse anharmonic oscillator is used for calculations of the energy of vibrational states, whereas a rigid rotator model is assumed for rotational levels [91].
-
Hydrogen: Electron impact cross sections for H2 are taken from the IST-Lisbon database of LXCat [92].
-
Nitrogen: Electron impact cross sections for N2 are taken from the IST-Lisbon database of LXCat [92].

This entry is adapted from the peer-reviewed paper 10.3390/molecules26123663
References
- Chang, S.; DesMarais, D.; Mack, R.; Miller, S.; Strathearn, G. Prebiotic Organic Syntheses and the Origin of Life, in Earth’s Earliest Atmospheres; Princeton University Press: Princeton, NJ, USA, 1983; pp. 53–92.
- Lazcano, A.; Miller, S.L. The origin and early evolution of life: Prebiotic chemistry, the pre-RNA world, and time. Cell 1996, 85, 793–798.
- Miller, S.L.; Cleaves, H.J. Prebiotic chemistry on the primitive Earth. Syst. Biol. 2006, 1, 1.
- Ehrenfreund, P.; Rasmussen, S.; Cleaves, J.; Chen, L. Experimentally tracing the key steps in the origin of life: The aromatic world. Astrobiology 2006, 6, 490–520.
- Chyba, C.; Sagan, C. Endogenous production, exogenous delivery and impact-shock synthesis of organic molecules: An inventory for the origins of life. Nature 1992, 355, 125–132.
- Bernstein, M. Prebiotic materials from on and off the early Earth. Philos. Trans. R. Soc. B Biol. Sci. 2006, 361, 1689–1702.
- Maurette, M.; Brack, A.; Kurat, G.; Perreau, M.; Engrand, C. Were micrometeorites a source of prebiotic molecules on the early Earth? Adv. Space Res. 1995, 15, 113–126.
- Ehrenfreund, P.; Irvine, W.; Becker, L.; Blank, J.; Brucato, J.; Colangeli, L.; Derenne, S.; Despois, D.; Dutrey, A.; Fraaije, H.; et al. Astrophysical and astrochemical insights into the origin of life. Rep. Prog. Phys. 2002, 65, 1427.
- Burchell, M.J. Panspermia today. Int. J. Astrobiol. 2004, 3, 73–80.
- Coulson, S. On panspermia and the survivability of micrometre-sized meteoroids within the Earth’s atmosphere. Int. J. Astrobiol. 2004, 3, 151–156.
- Flynn, G.; Keller, L.; Jacobsen, C.; Wirick, S. An assessment of the amount and types of organic matter contributed to the Earth by interplanetary dust. Adv. Space Res. 2004, 33, 57–66.
- Micca Longo, G.; Longo, S. Theoretical analysis of the atmospheric entry of sub-mm meteoroids of MgxCa1-xCO3 composition. Icarus 2018, 310, 194–202.
- Micca Longo, G.; Piccinni, V.; Longo, S. Evaluation of CaSO4 micrograins in the context of organic matter delivery: Thermochemistry and atmospheric entry. Int. J. Astrobiol. 2019, 18, 345–352.
- Micca Longo, G.; D’Elia, M.; Fonti, S.; Longo, S.; Mancarella, F.; Orofino, V. Kinetics of White Soft Minerals (WSMs) Decomposition under Conditions of Interest for Astrobiology: A Theoretical and Experimental Study. Geosciences 2019, 9, 101.
- Matrajt, G.; Messenger, S.; Brownlee, D.; Joswiak, D. Diverse forms of primordial organic matter identified in interplanetary dust particles. Meteorit. Planet. Sci. 2012, 47, 525–549.
- Yabuta, H.; Uesugi, M.; Naraoka, H.; Ito, M.; Kilcoyne, A.L.D.; Sandford, S.A.; Kitajima, F.; Mita, H.; Takano, Y.; Yada, T.; et al. X-ray absorption near edge structure spectroscopic study of Hayabusa category 3 carbonaceous particles. Earth Planets Space 2014, 66, 156.
- Dong, H.; Rech, J.A.; Jiang, H.; Sun, H.; Buck, B.J. Endolithic cyanobacteria in soil gypsum: Occurrences in Atacama (Chile), Mojave (United States), and Al-Jafr Basin (Jordan) Deserts. J. Geophys. Res. Biogeosci. 2007, 112.
- Gooding, J.L.; Wentworth, S.J.; Zolensky, M.E. Calcium carbonate and sulfate of possible extraterrestrial origin in the EETA 79001 meteorite. Geochim. Cosmochim. Acta 1988, 52, 909–915.
- Micca Longo, G.; Longo, S. The role of primordial atmosphere composition in organic matter delivery to early Earth. Rend. Lincei. Sci. Fis. E Nat. 2020, 31, 53–64.
- Miller, S.L. A Production of Amino Acids Under Possible Primitive Earth Conditions. Science 1953, 117, 528–529.
- Miller, S.L. Production of Some Organic Compounds under Possible Primitive Earth Conditions1. J. Am. Chem. Soc. 1955, 77, 2351–2361.
- Miller, S.L.; Urey, H.C. Organic Compound Synthes on the Primitive Eart. Science 1959, 130, 245–251.
- Oparin, A. Proischogdenie Zhizni; Moscovsky Robotchii: Moscow, Russia, 1924.
- Marshall, W.L. Hydrothermal synthesis of amino acids. Geochim. Cosmochim. Acta 1994, 58, 2099–2106.
- Lazcano, A.; Bada, J.L. The 1953 Stanley L. Miller Experiment: Fifty Years of Prebiotic Organic Chemistry. Orig. Life Evol. Biosph. 2003, 33, 235–242.
- Trainer, M.G.; Pavlov, A.A.; Curtis, D.B.; Mckay, C.P.; Worsnop, D.R.; Delia, A.E.; Toohey, D.W.; Toon, O.B.; Tolbert, M.A. Haze aerosols in the atmosphere of early Earth: Manna from heaven. Astrobiology 2004, 4, 409–419.
- Hasenkopf, C.A.; Freedman, M.A.; Beaver, M.R.; Toon, O.B.; Tolbert, M.A. Potential climatic impact of organic haze on early Earth. Astrobiology 2011, 11, 135–149.
- Balucani, N. Elementary reactions and their role in gas-phase prebiotic chemistry. Int. J. Mol. Sci. 2009, 10, 2304–2335.
- Bada, J.L. New insights into prebiotic chemistry from Stanley Miller’s spark discharge experiments. Chem. Soc. Rev. 2013, 42, 2186–2196.
- Haldane, J.B.S. Sciende and Human Life; Harper and Brothers: New York, NY, USA, 1933.
- Hart, M.H. The evolution of the atmosphere of the earth. Icarus 1978, 33, 23–39.
- Holland, H.D. Petrologic Studies: A Volume to Honor A.F. Buddington; Geological Society of America: New York, NY, USA, 1962; p. 447.
- Abelson, P.H. Chemical Events on the Primitive Earth. Proc. Natl. Acad. Sci. USA 1966, 55, 1365.
- Holland, H. The Chemical Evolution of the Atmosphere and Oceans; Princeton University Press: Princeton, NJ, USA, 1984.
- Kasting, J.F. Earth’s early atmosphere. Science 1993, 259, 920–926.
- Schlesinger, G.; Miller, S.L. Prebiotic synthesis in atmospheres containing CH4, CO, and CO2. J. Mol. Evol. 1983, 19, 376–382.
- Miyakawa, S.; Yamanashi, H.; Kobayashi, K.; Cleaves, H.J.; Miller, S.L. Prebiotic synthesis from CO atmospheres: Implications for the origins of life. Proc. Natl. Acad. Sci. USA 2002, 99, 14628.
- Cleaves, H.J.; Chalmers, J.H.; Lazcano, A.; Miller, S.L.; Bada, J.L. A Reassessment of Prebiotic Organic Synthesis in Neutral Planetary Atmospheres. Orig. Life Evol. Biosph. 2008, 38, 105–115.
- Zahnle, K.; Schaefer, L.; Fegley, B. Earth’s Earliest Atmospheres. Cold Spring Harb. Perspect. Biol. 2010, 2, a004895.
- Saitta, A.M.; Saija, F. Miller experiments in atomistic computer simulations. Proc. Natl. Acad. Sci. USA 2014, 111, 13768.
- Pietrucci, F.; Saitta, A.M. Formamide reaction network in gas phase and solution via a unified theoretical approach: Toward a reconciliation of different prebiotic scenarios. Proc. Natl. Acad. Sci. USA 2015, 112, 15030.
- Ferus, M.; Pietrucci, F.; Saitta, A.M.; Knížek, A.; Kubelík, P.; Ivanek, O.; Shestivska, V.; Civiš, S. Formation of nucleobases in a Miller–Urey reducing atmosphere. Proc. Natl. Acad. Sci. USA 2017, 114, 4306.
- Langmuir, I. Oscillations in ionized gases. Proc. Natl. Acad. Sci. USA 1928, 14, 627.
- Reid Thompson, W.; Henry, T.J.; Schwartz, J.M.; Khare, B.; Sagan, C. Plasma discharge in N2 + CH4 at low pressures: Experimental results and applications to Titan. Icarus 1991, 90, 57–73.
- Alcouffe, G.; Cavarroc, M.; Cernogora, G.; Ouni, F.; Jolly, A.; Boufendi, L.; Szopa, C. Capacitively coupled plasma used to simulate Titan’s atmospheric chemistry. Plasma Sources Sci. Technol. 2009, 19, 015008.
- Sciamma-O’Brien, E.; Ricketts, C.L.; Salama, F. The Titan Haze Simulation experiment on COSmIC: Probing Titan’s atmospheric chemistry at low temperature. Icarus 2014, 243, 325–336.
- Dubois, D.; Carrasco, N.; Jovanovic, L.; Vettier, L.; Gautier, T.; Westlake, J. Positive ion chemistry in an N2-CH4 plasma discharge: Key precursors to the growth of Titan tholins. Icarus 2020, 338, 113437.
- Köhn, C.; Dujko, S.; Chanrion, O.; Neubert, T. Streamer propagation in the atmosphere of Titan and other N2:CH4 mixtures compared to N2:O2 mixtures. Icarus 2019, 333, 294–305.
- Dubrovin, D.; Nijdam, S.; van Veldhuizen, E.M.; Ebert, U.; Yair, Y.; Price, C. Sprite discharges on Venus and Jupiter-like planets: A laboratory investigation. J. Geophys. Res. Space Phys. 2010, 115.
- Libby, W.F. Plasma chemistry. J. Vac. Sci. Technol. 1979, 16, 414–417.
- Gicquel, A.; Silva, F.; Hassouni, K. Diamond Growth Mechanisms in Various Environments. J. Electrochem. Soc. 2000, 147, 2218.
- Herrebout, D.; Bogaerts, A.; Yan, M.; Gijbels, R.; Goedheer, W.; Dekempeneer, E. One-dimensional fluid model for an rf methane plasma of interest in deposition of diamond-like carbon layers. J. Appl. Phys. 2001, 90, 570–579.
- Okita, A.; Suda, Y.; Oda, A.; Nakamura, J.; Ozeki, A.; Bhattacharyya, K.; Sugawara, H.; Sakai, Y. Effects of hydrogen on carbon nanotube formation in CH4/H2 plasmas. Carbon 2007, 45, 1518–1526.
- Physical Kinetics of Ionized Gases. In Fundamentals of Ionized Gases; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2011; Chapter 3; pp. 133–193.
- Meek, J.M.; Craggs, J.D. Electrical Breakdown of Gases; Oxford at The Clarendon Press: Oxford, UK, 1953.
- Von Engel, A. Electric Plasmas-Their Nature and Uses; Taylor and Francis, Ltd.: London, UK, 1983; 254p.
- Williams, E.R.; Geotis, S.G.; Bhattacharya, A. A radar study of the plasma and geometry of lightning. J. Atmos. Sci. 1989, 46, 1173–1185.
- Braglia, G. The diffusion and drift of electrons in gases a Monte-Carlo simulation. Phys. B+ C 1977, 92, 91–112.
- Schaefer, G.; Hui, P. The Monte Carlo flux method. J. Comput. Phys. 1990, 89, 1–30.
- Bogaerts, A.; Gijbels, R. Mathematical description of a direct current glow discharge in argon. Fresenius’ J. Anal. Chem. 1996, 355, 853–857.
- Biagi, S.F. Monte Carlo simulation of electron drift and diffusion in counting gases under the influence of electric and magnetic fields. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrometers Detect. Assoc. Equip. 1999, 421, 234–240.
- Longo, S. Monte Carlo models of electron and ion transport in non-equilibrium plasmas. Plasma Sources Sci. Technol. 2000, 9, 468–476.
- Loffhagen, D.; Winkler, R.; Donkó, Z. Boltzmann equation and Monte Carlo analysis of the spatiotemporal electron relaxation in nonisothermal plasmas. Eur. Phys. J. Appl. Phys. 2002, 18, 189–200.
- Hagelaar, G.J.M.; Pitchford, L.C. Solving the Boltzmann equation to obtain electron transport coefficients and rate coefficients for fluid models. Plasma Sources Sci. Technol. 2005, 14, 722–733.
- Capitelli, M.; Celiberto, R.; Colonna, G.; Esposito, F.; Gorse, C.; Hassouni, K.; Laricchiuta, A.; Longo, S. Fundamental Aspects of Plasma Chemical Physics: Kinetics; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2015; Volume 85.
- Vialetto, L.; Longo, S.; Diomede, P. Benchmark calculations for electron velocity distribution function obtained with Monte Carlo Flux simulations. Plasma Sources Sci. Technol. 2019, 28, 115015.
- Gordiets, B.F.; Osipov, A.I.; Stupochenko, E.V.; Shelepin, L.A. Vibrational Relaxation in Gases and Molecular Lasers. Sov. Phys. Uspekhi 1973, 15, 759–785.
- Capitelli, M.; Armenise, I.; Bruno, D.; Cacciatore, M.; Celiberto, R.; Colonna, G.; Pascale, O.D.; Diomede, P.; Esposito, F.; Gorse, C.; et al. Non-equilibrium plasma kinetics: A state-to-state approach. Plasma Sources Sci. Technol. 2007, 16, S30–S44.
- Laux, C.O.; Pierrot, L.; Gessman, R.J. State-to-state modeling of a recombining nitrogen plasma experiment. Chem. Phys. 2012, 398, 46–55.
- Kim, J.G.; Boyd, I.D. State-resolved master equation analysis of thermochemical nonequilibrium of nitrogen. Chem. Phys. 2013, 415, 237–246.
- Armenise, I.; Kustova, E. State-to-state models for CO2 molecules: From the theory to an application to hypersonic boundary layers. Chem. Phys. 2013, 415, 269–281.
- Kadochnikov, I.N.; Arsentiev, I.V. Kinetics of nonequilibrium processes in air plasma formed behind shock waves: State-to-state consideration. J. Phys. D: Appl. Phys. 2018, 51, 374001.
- Guerra, V.; Silva, T.; Ogloblina, P.; Grofulović, M.; Terraz, L.; da Silva, M.L.; Pintassilgo, C.D.; Alves, L.L.; Guaitella, O. The case for in situ resource utilisation for oxygen production on Mars by non-equilibrium plasmas. Plasma Sources Sci. Technol. 2017, 26, 11LT01.
- Pietanza, L.D.; Colonna, G.; D’Ammando, G.; Laricchiuta, A.; Capitelli, M. Vibrational excitation and dissociation mechanisms of CO2 under non-equilibrium discharge and post-discharge conditions. Plasma Sources Sci. Technol. 2015, 24, 042002.
- Rusanov, V.D.; Fridman, A.A.; Sholin, G.V. The physics of a chemically active plasma with nonequilibrium vibrational excitation of molecules. Sov. Phys. Uspekhi 1981, 24, 447–474.
- Viegas, P.; van de Sanden, M.C.M.; Longo, S.; Diomede, P. Validation of the Fokker–Planck Approach to Vibrational Kinetics in CO2 Plasma. J. Phys. Chem. C 2019, 123, 22823–22831.
- Longo, S.; van de Sanden, M.C.M.; Diomede, P. Fokker–Planck equation for chemical reactions in plasmas. Rend. Lincei. Sci. Fis. E Nat. 2019, 30, 25–30.
- Diomede, P.; van de Sanden, M.C.M.; Longo, S. Vibrational Kinetics in Plasma as a Functional Problem: A Flux-Matching Approach. J. Phys. Chem. A 2018, 122, 7918–7923.
- Petrović, Z.L.; Dujko, S.; Marić, D.; Malović, G.; Nikitović, Ž.; Šašić, O.; Jovanović, J.; Stojanović, V.; Radmilović-Radjenović, M. Measurement and interpretation of swarm parameters and their application in plasma modelling. J. Phys. D Appl. Phys. 2009, 42.
- Tejero-del-Caz, A.; Guerra, V.; Gonçalves, D.; da Silva, M.L.; Marques, L.; Pinhão, N.; Pintassilgo, C.D.; Alves, L.L. The LisbOn KInetics Boltzmann solver. Plasma Sources Sci. Technol. 2019, 28, 043001.
- Stephens, J. A multi-term Boltzmann equation benchmark of electron-argon cross-sections for use in low temperature plasma models. J. Phys. D Appl. Phys. 2018, 51, 125203.
- Rabie, M.; Franck, C.M. METHES: A Monte Carlo collision code for the simulation of electron transport in low temperature plasmas. Comput. Phys. Comm. 2016, 203, 268–277.
- Vialetto, L.; Viegas, P.; Longo, S.; Diomede, P. Benchmarking of Monte Carlo Flux simulations of electrons in CO2. Plasma Sources Sci. Technol. 2020.
- McCollom, T.M. Miller-Urey and beyond: What have we learned about prebiotic organic synthesis reactions in the past 60 years? Annu. Rev. Earth Planet. Sci. 2013, 41, 207–229.
- Scherer, S.; Wollrab, E.; Codutti, L.; Carlomagno, T.; da Costa, S.G.; Volkmer, A.; Bronja, A.; Schmitz, O.J.; Ott, A. Chemical analysis of a “Miller-type” complex prebiotic broth. Orig. Life Evol. Biosph. 2017, 47, 381–403.
- Fowler, R.H. XXIII. Statistical equilibrium with special reference to the mechanism of ionization by electronic impacts. Lond. Edinb. Dublin Philos. Mag. J. Sci. 1924, 47, 257–277.
- Magboltz - Transport of Electrons in Gas Mixtures. Available online: http://magboltz.web.cern.ch/magboltz/ (accessed on 2 August 2019).
- Hayashi Database. Available online: www.lxcat.net (accessed on 18 December 2020).
- Biagi Database. Available online: www.lxcat.net (accessed on 18 December 2020).
- Vialetto, L.; Guerra, V.; Diomede, P.; Longo, S.; Ben Moussa, A.; van Dijk, J.; Alves, L.L. Effect of anisotropic scattering on electron transport parameters in CO. Plasma Sources Sci. Technol. 2021. in preparation.
- Capitelli, M.; Ferreira, C.M.; Gordiets, B.F.; Osipov, A.I. Plasma Kinetics in Atmospheric Gases; Springer Science & Business Media: Berlin/Heidelberg, Germany, 2013; Volume 31.
- IST-Lisbon Database. Available online: www.lxcat.net (accessed on 18 December 2020).