Takotsubo syndrome (TTS) is identified as an acute severe ventricular systolic dysfunction, which is usually characterized by reversible and transient akinesia of walls of the ventricle in the absence of a significant obstructive coronary artery disease (CAD). Patients present with chest pain, ST-segment elevation or ischemia signs on ECG and increased troponin, similar to myocardial infarction. Currently, the known mechanisms associated with the development of TTS include elevated levels of circulating plasma catecholamines and their metabolites, coronary microvascular dysfunction, sympathetic hyperexcitability, inflammation, estrogen deficiency, spasm of the epicardial coronary vessels, genetic predisposition and thyroidal dysfunction.
- Takotsubo syndrome
- pathophysiological mechanism
- human-induced pluripotent stem cell-derived cardiomyocytes
- catecholamines
- precision medicine
1. Introduction
2. Experimental Models of TTS
2.1. Animal Models and Mechanistic Studies
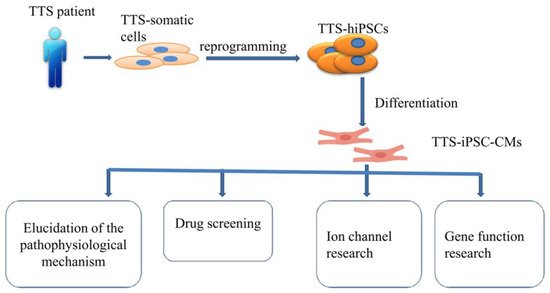
2.2. Human Cardiomyocytes Derived from Induced Pluripotent Stem Cells (hiPSC-CMs) Models
2.3. Other Models
| Model | Method | Main Finding | |
|---|---|---|---|
| Animal model | Rats [20][22][23][26][27][28][29] | Immobilization (IMO) | (1) The activation of β1 adrenergic receptors in the heart and the activation of α1 adrenergic receptors in the aorta were the primary cause of TTS (2) The reduction of estrogen may interfere with coronary microcirculation and may be involved in the primary cause of TTS by indirect action on the nervous system and by direct action on the heart (3) α2AR/Gi-dependent signaling attenuated myosin-binding protein-C(MyBP-C) phosphorylation and contractility in the anterior wall (AW) through an epinephrine surge in TTS rats |
| Ovariectomized (OVX) and estradiol-supplemented ovariectomized female rats [19][21] | Immobilization (IMO) | (1) The reduction of LV contractility and the increase of heart rate in response to emotional stress were attenuated by supplement of estradiol in the ovariectomized rats. (2) Emotional stress and a surge of catecholamine upregulated heme oxygenase-1 (HO-1) |
|
| Cynomolgus monkeys [33] | Intravenous infusion of epinephrine overdose | LV dysfunction with apical ballooning and wall motion abnormalities | |
| Rabbits [34] | Vagal stimulation | The cardiac lesions related to ventricular arrhythmias were involved in the basal portion, mitral valve, and papillary muscles but not the apex | |
| Mice [25][36][47] | A single dose injection of isoprenaline | (1) Lipotoxicity was closely related to catecholamine-induced myocardial dysfunction, including neurogenic stunning, metabolic stunning, and electrophysiological stunning (2) ISO reduced GLS and circumferential (GCS) strains of males and females |
|
| Rats [30][35][38][39][40][41][42][43][44][46] | Isoprenaline (ISO) | (1) TTS rats had significantly lower left ventricular end-diastolic pressure and significantly better estimates of cardiac function. (2) Its apical perfusion was not impaired in the early stage of TTS (3) ISO significantly increased the levels of reactive oxygen species (ROS) in the setting of TTS (4) Early treatment with isoflurane could reduce LV dyskinesia and improve the survival rate of experimental TTS (5) GPER, azelnidipine, Tempol and amlodipine also played a protective role for TTS |
|
| Rats [45] | Epinephrine | GPER played a protective role against TTS | |
| hiPSC-CMs models | hiPSC-CMs models [14] | Isoprenaline | Estradiol had protective effects against catecholamine excess and hence reduction in estrogen level may increase the risk of acquired long QT syndrome in TTC |
| hiPSC-CMs models [45] | Epinephrine | Knockdown of GPER by siRNA abolished E2 effects on increasing ICa-L and action potential duration in the stress state | |
| hiPSC-CMs models [62] | Epinephrine | High concentrations of epinephrine inhibited the depolarization rate in hiPSC-CMs, the duration of action potentials and induced arrhythmia events while the effect of epinephrine was attenuated by alpha-adrenergic receptor blockers-phentolamine | |
| TTS-iPSC-CMs [63] | The β-adrenergic signaling, including cAMP response and cAMP-dependent PKA activity, was increased in TTS-iPSC-CMs | ||
| Other cells model | H9C2 [46] | Isoproterenol | Pretreatment with Tempolcould reduce the production of reactive oxygen species and the deposition of lipid droplets and protect mitochondrial function by reducing mitochondrial swelling |
| Computational model [71] | Three potential dominant mechanisms are related to the effects of β-adrenergic stimulation |
This entry is adapted from the peer-reviewed paper 10.3390/ijms23041951
References
- Cramer, M.J.; De Boeck, B.; Melman, P.G.; Sieswerda, G.J. The ‘broken heart’ syndrome: What can be learned from the tears and distress? Neth. Heart J. 2007, 15, 283–285.
- Sharkey, S.W.; Windenburg, D.C.; Lesser, J.R.; Maron, M.S.; Hauser, R.G.; Lesser, J.N.; Haas, T.S.; Hodges, J.S.; Maron, B.J. Natural history and expansive clinical profile of stress (tako-tsubo) cardiomyopathy. J. Am. Coll. Cardiol. 2010, 55, 333–341.
- Topal, Y.; Topal, H.; Dogan, C.; Tiryaki, S.B.; Biteker, M. Takotsubo (stress) cardiomyopathy in childhood. Eur. J. Pediatr. 2020, 179, 619–625.
- Dote, K.; Sato, H.; Tateishi, H.; Uchida, T.; Ishihara, M. Myocardial stunning due to simultaneous multivessel coronary spasms: A review of 5 cases. J. Cardiol. 1991, 21, 203–214.
- Lyon, A.R.; Bossone, E.; Schneider, B.; Sechtem, U.; Citro, R.; Underwood, S.R.; Sheppard, M.N.; Figtree, G.A.; Parodi, G.; Akashi, Y.J.; et al. Current state of knowledge on Takotsubo syndrome: A Position Statement from the Taskforce on Takotsubo Syndrome of the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2016, 18, 8–27.
- Prasad, A.; Lerman, A.; Rihal, C.S. Apical ballooning syndrome (Tako-Tsubo or stress cardiomyopathy): A mimic of acute myocardial infarction. Am. Heart J. 2008, 155, 408–417.
- Ancona, F.; Bertoldi, L.F.; Ruggieri, F.; Cerri, M.; Magnoni, M.; Beretta, L.; Cianflone, D.; Camici, P.G. Takotsubo cardiomyopathy and neurogenic stunned myocardium: Similar albeit different. Eur. Heart J. 2016, 37, 2830–2832.
- Madias, J.E. “Neurogenic stress cardiomyopathy in heart donors” is a form of Takotsubo syndrome. Int. J. Cardiol. 2015, 184, 612–613.
- Mohamedali, B.; Bhat, G.; Zelinger, A. Frequency and pattern of left ventricular dysfunction in potential heart donors: Implications regarding use of dysfunctional hearts for successful transplantation. J. Am. Coll. Cardiol. 2012, 60, 235–236.
- Moussouttas, M.; Mearns, E.; Walters, A.; DeCaro, M. Plasma Catecholamine Profile of Subarachnoid Hemorrhage Patients with Neurogenic Cardiomyopathy. Cerebrovasc. Dis. Extra 2015, 5, 57–67.
- Tavazzi, G.; Zanierato, M.; Via, G.; Iotti, G.A.; Procaccio, F. Are Neurogenic Stress Cardiomyopathy and Takotsubo Different Syndromes With Common Pathways?: Etiopathological Insights on Dysfunctional Hearts. JACC Heart Fail. 2017, 5, 940–942.
- Aweimer, A.; El-Battrawy, I.; Akin, I.; Borggrefe, M.; Mugge, A.; Patsalis, P.C.; Urban, A.; Kummer, M.; Vasileva, S.; Stachon, A.; et al. Abnormal thyroid function is common in takotsubo syndrome and depends on two distinct mechanisms: Results of a multicentre observational study. J. Intern. Med. 2021, 289, 675–687.
- Naegele, M.; Flammer, A.J.; Enseleit, F.; Roas, S.; Frank, M.; Hirt, A.; Kaiser, P.; Cantatore, S.; Templin, C.; Frohlich, G.; et al. Endothelial function and sympathetic nervous system activity in patients with Takotsubo syndrome. Int. J. Cardiol. 2016, 224, 226–230.
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Schunemann, J.D.; Sattler, K.; Buljubasic, F.; Patocskai, B.; Li, X.; Yucel, G.; Lang, S.; et al. Estradiol protection against toxic effects of catecholamine on electrical properties in human-induced pluripotent stem cell derived cardiomyocytes. Int. J. Cardiol. 2018, 254, 195–202.
- Scally, C.; Abbas, H.; Ahearn, T.; Srinivasan, J.; Mezincescu, A.; Rudd, A.; Spath, N.; Yucel-Finn, A.; Yuecel, R.; Oldroyd, K.; et al. Myocardial and Systemic Inflammation in Acute Stress-Induced (Takotsubo) Cardiomyopathy. Circulation 2019, 139, 1581–1592.
- Paur, H.; Wright, P.T.; Sikkel, M.B.; Tranter, M.H.; Mansfield, C.; O’Gara, P.; Stuckey, D.J.; Nikolaev, V.O.; Diakonov, I.; Pannell, L.; et al. High levels of circulating epinephrine trigger apical cardiodepression in a beta2-adrenergic receptor/Gi-dependent manner: A new model of Takotsubo cardiomyopathy. Circulation 2012, 126, 697–706.
- Orphanou, N.; Eftychiou, C.; Papasavvas, E.; Ioannides, M.; Avraamides, P. Syncope in a hypertrophic heart at a wedding party: Can happiness break a thick heart? Takotsubo cardiomyopathy complicated with left ventricular outflow tract obstruction in a hypertrophic heart. Oxf Med. Case Rep. 2020, 2020, omaa036.
- Bonacchi, M.; Vannini, A.; Harmelin, G.; Batacchi, S.; Bugetti, M.; Sani, G.; Peris, A. Inverted-Takotsubo cardiomyopathy: Severe refractory heart failure in poly-trauma patients saved by emergency extracorporeal life support. Interact. Cardiovasc. Thorac. Surg. 2015, 20, 365–371.
- Ueyama, T.; Kawabe, T.; Hano, T.; Tsuruo, Y.; Ueda, K.; Ichinose, M.; Kimura, H.; Yoshida, K. Upregulation of heme oxygenase-1 in an animal model of Takotsubo cardiomyopathy. Circ. J. 2009, 73, 1141–1146.
- Ishikura, F.; Takano, Y.; Ueyama, T. Amlodipine has a preventive effect on temporal left ventricular hypokinesia after emotional stress compared with an angiotensin II receptor blocker. J. Med. Ultrason. (2001) 2013, 40, 3–7.
- Ueyama, T.; Ishikura, F.; Matsuda, A.; Asanuma, T.; Ueda, K.; Ichinose, M.; Kasamatsu, K.; Hano, T.; Akasaka, T.; Tsuruo, Y.; et al. Chronic estrogen supplementation following ovariectomy improves the emotional stress-induced cardiovascular responses by indirect action on the nervous system and by direct action on the heart. Circ. J. 2007, 71, 565–573.
- Kuroda, R.; Shintani-Ishida, K.; Unuma, K.; Yoshida, K. Immobilization Stress With alpha2-Adrenergic Stimulation Induces Regional and Transient Reduction of Cardiac Contraction Through Gi Coupling in Rats. Int. Heart J. 2015, 56, 537–543.
- Ueyama, T.; Yamamoto, Y.; Ueda, K.; Kawabe, T.; Hano, T.; Ito, T.; Tsuruo, Y.; Ichinose, M.; Yoshida, K. Cardiac and vascular gene profiles in an animal model of takotsubo cardiomyopathy. Heart Vessel. 2011, 26, 321–337.
- Ueyama, T. Emotional stress-induced Tako-tsubo cardiomyopathy: Animal model and molecular mechanism. Ann. N. Y. Acad. Sci. 2004, 1018, 437–444.
- Zhang, H.; Sun, Y.; Liu, X.; Yang, Y.; Sun, T.; Krittanawong, C.; El-Am, E.A.; Liu, G.; Yang, J.; Ma, N. Speckle tracking echocardiography in early detection of myocardial injury in a rat model with stress cardiomyopathy. Med. Ultrason. 2019, 21, 441–448.
- Takano, Y.; Ueyama, T.; Ishikura, F. Azelnidipine, unique calcium channel blocker could prevent stress-induced cardiac dysfunction like alpha.beta blocker. J. Cardiol. 2012, 60, 18–22.
- Ueyama, T.; Kasamatsu, K.; Hano, T.; Tsuruo, Y.; Ishikura, F. Catecholamines and estrogen are involved in the pathogenesis of emotional stress-induced acute heart attack. Ann. N. Y. Acad. Sci. 2008, 1148, 479–485.
- Ueyama, T.; Kasamatsu, K.; Hano, T.; Yamamoto, K.; Tsuruo, Y.; Nishio, I. Emotional stress induces transient left ventricular hypocontraction in the rat via activation of cardiac adrenoceptors: A possible animal model of ‘tako-tsubo’ cardiomyopathy. Circ. J. 2002, 66, 712–713.
- Kvetnansky, R.; Pacak, K.; Fukuhara, K.; Viskupic, E.; Hiremagalur, B.; Nankova, B.; Goldstein, D.S.; Sabban, E.L.; Kopin, I.J. Sympathoadrenal system in stress. Interaction with the hypothalamic-pituitary-adrenocortical system. Ann. N. Y. Acad. Sci. 1995, 771, 131–158.
- Kolodzinska, A.; Czarzasta, K.; Szczepankiewicz, B.; Budnik, M.; Glowczynska, R.; Fojt, A.; Ilczuk, T.; Krasuski, K.; Borodzicz, S.; Cudnoch-Jedrzejewska, A.; et al. Isoprenaline induced Takotsubo syndrome: Histopathological analyses of female rat hearts. Cardiol. J. 2020.
- Ueyama, T.; Hano, T.; Kasamatsu, K.; Yamamoto, K.; Tsuruo, Y.; Nishio, I. Estrogen attenuates the emotional stress-induced cardiac responses in the animal model of Tako-tsubo (Ampulla) cardiomyopathy. J. Cardiovasc. Pharmacol. 2003, 42 (Suppl. S1), S117–S119.
- Wright, P.T.; Bhogal, N.K.; Diakonov, I.; Pannell, L.M.K.; Perera, R.K.; Bork, N.I.; Schobesberger, S.; Lucarelli, C.; Faggian, G.; Alvarez-Laviada, A.; et al. Cardiomyocyte Membrane Structure and cAMP Compartmentation Produce Anatomical Variation in beta2AR-cAMP Responsiveness in Murine Hearts. Cell Rep. 2018, 23, 459–469.
- Izumi, Y.; Okatani, H.; Shiota, M.; Nakao, T.; Ise, R.; Kito, G.; Miura, K.; Iwao, H. Effects of metoprolol on epinephrine-induced takotsubo-like left ventricular dysfunction in non-human primates. Hypertens. Res. 2009, 32, 339–346.
- Takato, T.; Ashida, T.; Seko, Y.; Fujii, J.; Kawai, S. Ventricular tachyarrhythmia-related basal cardiomyopathy in rabbits with vagal stimulation--a novel experimental model for inverted Takotsubo-like cardiomyopathy. J. Cardiol. 2010, 56, 85–90.
- Sachdeva, J.; Dai, W.; Kloner, R.A. Functional and histological assessment of an experimental model of Takotsubo’s cardiomyopathy. J. Am. Heart Assoc. 2014, 3, e000921.
- Shao, Y.; Redfors, B.; Stahlman, M.; Tang, M.S.; Miljanovic, A.; Mollmann, H.; Troidl, C.; Szardien, S.; Hamm, C.; Nef, H.; et al. A mouse model reveals an important role for catecholamine-induced lipotoxicity in the pathogenesis of stress-induced cardiomyopathy. Eur. J. Heart Fail. 2013, 15, 9–22.
- Kolodzinska, A.; Czarzasta, K.; Szczepankiewicz, B.; Glowczynska, R.; Fojt, A.; Ilczuk, T.; Budnik, M.; Krasuski, K.; Folta, M.; Cudnoch-Jedrzejewska, A.; et al. Toll-like receptor expression and apoptosis morphological patterns in female rat hearts with takotsubo syndrome induced by isoprenaline. Life Sci. 2018, 199, 112–121.
- Godsman, N.; Kohlhaas, M.; Nickel, A.; Cheyne, L.; Mingarelli, M.; Schweiger, L.; Hepburn, C.; Munts, C.; Welch, A.; Delibegovic, M.; et al. Metabolic alterations in a rat model of Takotsubo syndrome. Cardiovasc. Res. 2021.
- Qi, C.; Shao, Y.; Liu, X.; Wang, D.; Li, X. The cardioprotective effects of icariin on the isoprenaline-induced takotsubo-like rat model: Involvement of reactive oxygen species and the TLR4/NF-kappaB signaling pathway. Int. Immunopharmacol. 2019, 74, 105733.
- Mao, S.; Luo, X.; Li, Y.; He, C.; Huang, F.; Su, C. Role of PI3K/AKT/mTOR Pathway Associated Oxidative Stress and Cardiac Dysfunction in Takotsubo Syndrome. Curr. Neurovasc. Res. 2020, 17, 35–43.
- Perez-Trevino, P.; Sepulveda-Leal, J.; Altamirano, J. Simultaneous assessment of calcium handling and contractility dynamics in isolated ventricular myocytes of a rat model of post-acute isoproterenol-induced cardiomyopathy. Cell Calcium. 2020, 86, 102138.
- Willis, B.C.; Salazar-Cantu, A.; Silva-Platas, C.; Fernandez-Sada, E.; Villegas, C.A.; Rios-Argaiz, E.; Gonzalez-Serrano, P.; Sanchez, L.A.; Guerrero-Beltran, C.E.; Garcia, N.; et al. Impaired oxidative metabolism and calcium mishandling underlie cardiac dysfunction in a rat model of post-acute isoproterenol-induced cardiomyopathy. Am. J. Physiol. Heart Circ. Physiol. 2015, 308, H467–H477.
- Oras, J.; Redfors, B.; Ali, A.; Alkhoury, J.; Seeman-Lodding, H.; Omerovic, E.; Ricksten, S.E. Early treatment with isoflurane attenuates left ventricular dysfunction and improves survival in experimental Takotsubo. Acta Anaesthesiol. Scand. 2017, 61, 399–407.
- Zhang, Z.; Jin, S.; Teng, X.; Duan, X.; Chen, Y.; Wu, Y. Hydrogen sulfide attenuates cardiac injury in takotsubo cardiomyopathy by alleviating oxidative stress. Nitric Oxide 2017, 67, 10–25.
- Fu, L.; Zhang, H.; Ong’achwa Machuki, J.; Zhang, T.; Han, L.; Sang, L.; Wu, L.; Zhao, Z.; James Turley, M.; Hu, X.; et al. GPER mediates estrogen cardioprotection against epinephrine-induced stress. J. Endocrinol. 2021, 249, 209–222.
- Qi, C.; Liu, X.; Xiong, T.; Wang, D. Tempol prevents isoprenaline-induced takotsubo syndrome via the reactive oxygen species/mitochondrial/anti-apoptosis /p38 MAPK pathway. Eur. J. Pharmacol. 2020, 886, 173439.
- Walsh-Wilkinson, E.; Arsenault, M.; Couet, J. Segmental analysis by speckle-tracking echocardiography of the left ventricle response to isoproterenol in male and female mice. PeerJ 2021, 9, e11085.
- Herraez, P.; Espinosa de los Monteros, A.; Fernandez, A.; Edwards, J.F.; Sacchini, S.; Sierra, E. Capture myopathy in live-stranded cetaceans. Vet. J. 2013, 196, 181–188.
- Harthoorn, A.M.; van der Walt, K.; Young, E. Possible therapy for capture myopathy in captured wild animals. Nature 1974, 247, 577.
- El-Battrawy, I.; Zhao, Z.; Lan, H.; Cyganek, L.; Tombers, C.; Li, X.; Buljubasic, F.; Lang, S.; Tiburcy, M.; Zimmermann, W.H.; et al. Electrical dysfunctions in human-induced pluripotent stem cell-derived cardiomyocytes from a patient with an arrhythmogenic right ventricular cardiomyopathy. Europace 2018, 20, f46–f56.
- El-Battrawy, I.; Besler, J.; Ansari, U.; Liebe, V.; Schimpf, R.; Tulumen, E.; Rudic, B.; Lang, S.; Odening, K.; Cyganek, L.; et al. Long-term follow-up of implantable cardioverter-defibrillators in Short QT syndrome. Clin. Res. Cardiol. 2019, 108, 1140–1146.
- El-Battrawy, I.; Albers, S.; Cyganek, L.; Zhao, Z.; Lan, H.; Li, X.; Xu, Q.; Kleinsorge, M.; Huang, M.; Liao, Z.; et al. A cellular model of Brugada syndrome with SCN10A variants using human-induced pluripotent stem cell-derived cardiomyocytes. Europace 2019, 21, 1410–1421.
- El-Battrawy, I.; Muller, J.; Zhao, Z.; Cyganek, L.; Zhong, R.; Zhang, F.; Kleinsorge, M.; Lan, H.; Li, X.; Xu, Q.; et al. Studying Brugada Syndrome With an SCN1B Variants in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2019, 7, 261.
- Lan, H.; Xu, Q.; El-Battrawy, I.; Zhong, R.; Li, X.; Lang, S.; Cyganek, L.; Borggrefe, M.; Zhou, X.; Akin, I. Ionic Mechanisms of Disopyramide Prolonging Action Potential Duration in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes From a Patient With Short QT Syndrome Type 1. Front. Pharmacol. 2020, 11, 554422.
- El-Battrawy, I.; Lan, H.; Cyganek, L.; Zhao, Z.; Li, X.; Buljubasic, F.; Lang, S.; Yucel, G.; Sattler, K.; Zimmermann, W.H.; et al. Modeling Short QT Syndrome Using Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes. J. Am. Heart Assoc. 2018, 7.
- Buljubasic, F.; El-Battrawy, I.; Lan, H.; Lomada, S.K.; Chatterjee, A.; Zhao, Z.; Li, X.; Zhong, R.; Xu, Q.; Huang, M.; et al. Nucleoside Diphosphate Kinase B Contributes to Arrhythmogenesis in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes from a Patient with Arrhythmogenic Right Ventricular Cardiomyopathy. J. Clin. Med. 2020, 9, 486.
- Zhao, Z.; Li, X.; El-Battrawy, I.; Lan, H.; Zhong, R.; Xu, Q.; Huang, M.; Liao, Z.; Lang, S.; Zimmermann, W.H.; et al. Drug Testing in Human-Induced Pluripotent Stem Cell-Derived Cardiomyocytes From a Patient With Short QT Syndrome Type 1. Clin. Pharmacol. Ther. 2019, 106, 642–651.
- Yucel, G.; Zhao, Z.; El-Battrawy, I.; Lan, H.; Lang, S.; Li, X.; Buljubasic, F.; Zimmermann, W.H.; Cyganek, L.; Utikal, J.; et al. Lipopolysaccharides induced inflammatory responses and electrophysiological dysfunctions in human-induced pluripotent stem cell derived cardiomyocytes. Sci. Rep. 2017, 7, 2935.
- Fan, X.; Yang, G.; Kowitz, J.; Duru, F.; Saguner, A.M.; Akin, I.; Zhou, X.; El-Battrawy, I. Preclinical short QT syndrome models: Studying the phenotype and drug-screening. Europace 2021.
- El-Battrawy, I.; Lan, H.; Cyganek, L.; Maywald, L.; Zhong, R.; Zhang, F.; Xu, Q.; Lee, J.; Duperrex, E.; Hierlemann, A.; et al. Deciphering the pathogenic role of a variant with uncertain significance for short QT and Brugada syndromes using gene-edited human-induced pluripotent stem cell-derived cardiomyocytes and preclinical drug screening. Clin. Transl. Med. 2021, 11, e646.
- Lach, R.; Schön, J.; Krolopp, T.; Arndt, S.; Langer, B.; Grellmann, W. Depth-Sensing Macroindentation Test and Stepped Isothermal Method–Accelerated Assessment of the Local Retardation Behaviour of Thermoplastic Polymers. Macromol. Symp. 2016, 366, 60–65.
- Huang, M.; Fan, X.; Yang, Z.; Cyganek, L.; Li, X.; Yuecel, G.; Lan, H.; Li, Y.; Wendel, A.; Lang, S.; et al. Alpha 1-adrenoceptor signalling contributes to toxic effects of catecholamine on electrical properties in cardiomyocytes. Europace 2021, 23, 1137–1148.
- Borchert, T.; Hubscher, D.; Guessoum, C.I.; Lam, T.D.; Ghadri, J.R.; Schellinger, I.N.; Tiburcy, M.; Liaw, N.Y.; Li, Y.; Haas, J.; et al. Catecholamine-Dependent beta-Adrenergic Signaling in a Pluripotent Stem Cell Model of Takotsubo Cardiomyopathy. J. Am. Coll. Cardiol. 2017, 70, 975–991.
- El-Battrawy, I.; Lang, S.; Ansari, U.; Tulumen, E.; Schramm, K.; Fastner, C.; Zhou, X.; Hoffmann, U.; Borggrefe, M.; Akin, I. Prevalence of malignant arrhythmia and sudden cardiac death in takotsubo syndrome and its management. Europace 2018, 20, 843–850.
- Ebert, A.; Joshi, A.U.; Andorf, S.; Dai, Y.; Sampathkumar, S.; Chen, H.; Li, Y.; Garg, P.; Toischer, K.; Hasenfuss, G.; et al. Proteasome-Dependent Regulation of Distinct Metabolic States During Long-Term Culture of Human iPSC-Derived Cardiomyocytes. Circ. Res. 2019, 125, 90–103.
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2017, 7, 8590.
- Sun, X.; Nunes, S.S. Maturation of Human Stem Cell-derived Cardiomyocytes in Biowires Using Electrical Stimulation. J. Vis. Exp. 2017.
- Ruan, J.L.; Tulloch, N.L.; Razumova, M.V.; Saiget, M.; Muskheli, V.; Pabon, L.; Reinecke, H.; Regnier, M.; Murry, C.E. Mechanical Stress Conditioning and Electrical Stimulation Promote Contractility and Force Maturation of Induced Pluripotent Stem Cell-Derived Human Cardiac Tissue. Circulation 2016, 134, 1557–1567.
- Herron, T.J.; Rocha, A.M.; Campbell, K.F.; Ponce-Balbuena, D.; Willis, B.C.; Guerrero-Serna, G.; Liu, Q.; Klos, M.; Musa, H.; Zarzoso, M.; et al. Extracellular Matrix-Mediated Maturation of Human Pluripotent Stem Cell-Derived Cardiac Monolayer Structure and Electrophysiological Function. Circ. Arrhythm. Electrophysiol. 2016, 9, e003638.
- Nunes, S.S.; Miklas, J.W.; Liu, J.; Aschar-Sobbi, R.; Xiao, Y.; Zhang, B.; Jiang, J.; Masse, S.; Gagliardi, M.; Hsieh, A.; et al. Biowire: A platform for maturation of human pluripotent stem cell-derived cardiomyocytes. Nat. Methods 2013, 10, 781–787.
- Land, S.; Niederer, S.A.; Louch, W.E.; Roe, A.T.; Aronsen, J.M.; Stuckey, D.J.; Sikkel, M.B.; Tranter, M.H.; Lyon, A.R.; Harding, S.E.; et al. Computational modeling of Takotsubo cardiomyopathy: Effect of spatially varying beta-adrenergic stimulation in the rat left ventricle. Am. J. Physiol. Heart Circ. Physiol. 2014, 307, H1487–H1496.