Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Developmental Biology
Prenatal hypoxia is a common complication in pregnancy, developing from various causes. Prenatal hypoxia during the prenatal period can interfere with the developmental trajectory and lead to developing hypertension in adulthood. Prenatal hypoxia is often associated with intrauterine growth restriction that interferes with metabolism and can lead to multilevel changes.
- prenatal hypoxia
- prenatal programming
- cardiovascular system
1. Introduction
The cardiovascular system is a dynamic system that can adapt to adverse conditions to maintain and satisfy homeostasis in the organism. Prolonged cardiovascular adaptations can result in the development of hypertension and other cardiovascular diseases in adulthood; however, hypertension can have its origins in the prenatal period when the cardiovascular system develops structurally and functionally [1]. This phenomenon observed in animals and humans is described as prenatal programming [2,3].
Prenatal programming is defined as a response to adverse factors acting during a critical prenatal period, leading to changes in the developmental trajectory with a permanent effect on the offspring’s adult phenotype [3]. Insults during the prenatal period lead to permanent structural or functional changes of the tissues and organs [3]. In general, if the prenatal factor acts during the early phase of an organ’s development, it leads to structural defects. In contrast, the action during the later phases of development affects functions [4]. The effect of prenatal insults on the foetus depends not only on the developmental stage but also on the type of insult. Consequently, some types of insults, such as various agents (warfarin, thalidomide, tetracycline, alcohol) and environmental factors of chemical (toxic metals), physical (ionising radiation) or biological origin (infection diseases), can disrupt the normal in utero development of the foetus and increase the risk of congenital disabilities, malformations, and in some cases, even death of the developing foetus [5].
Moderate effects on the foetus include high or low food availability, oxygen deficiency (hypoxia), maternal obesity, inadequate prenatal care, maternal stress, and maternal chronic diseases. The consequences of these factors can be identified during pregnancy screening and immediately after birth because they are often associated with a reduction in birth weight and asymmetric organ growth [6]. According to Barker’s theory, neonates with reduced birth weight have a higher incidence of stroke and coronary heart disease in adulthood [7]. A reduction in birth weight of neonates is the most accessible marker of poor in utero development. Indeed, many studies have recently explored an association between intrauterine growth restriction and an increased postnatal risk of cardiovascular diseases [8,9,10].
However, prenatal insults do not necessarily have to be manifested by low birth weight (Table 1) and still can lead to the programming of diseases in adulthood. This phenomenon is observed after exposure to prenatal hypoxia, which is a common complication in gravidity. In this case, neonates are born seemingly healthy, avoiding the early screening of diseases; however, due to prenatal hypoxia, the foetus has impaired endothelial function and undergoes oxidative stress [2,9,11], morphological changes in the heart and blood vessels [2,12] and changes in the activity of the autonomic nervous system [13,14], which is one of the key factors in the development of hypertension [15]. Prenatal hypoxia contributes to the development of hypertension, ischemic heart disease, coronary heart disease, heart failure, metabolic syndrome, and increased susceptibility to ischemic injury in humans [16,17].
Table 1. The effect of prenatal hypoxia on birth body weight.
| Oxygen | Duration; Time | Animal Model | Birth Body Weight | Ref. |
|---|---|---|---|---|
| 6.5% | 1–20 ED; 8 h per day: 80 s hypoxia and 120 s normoxia; 18 cycles per hour | Sprague Dawley rats | ↓ | [18] |
| 7% | 13–14 ED; 3 h | Wistar rats | ↓ | [19] |
| 7% | 18 ED; 3 h | Wistar rats | = | [20] |
| 9% | 15–21 ED; 6 h per day | Sprague Dawley rats | = | [21] |
| 9.5–10% | 12, 24, 48, 120 h immediately prior to delivery at term | Sprague Dawley rats | ↓ | [22] |
| 10% | 5–19 ED | Sprague Dawley rats | ↓ | [23] |
| 10% | 5–20 ED | Sprague Dawley rats | ↓ | [13,24,25,26,27] |
| 10% | 15–20 ED | Wistar rats | ↓ | [28,29] |
| 10% | from 121 ED–NA | sheep | = | [30] |
| 10 ± 0.5% | 5–20 ED | Sprague Dawley rats | ↓ | [31] |
| 10.5% | 15–20 ED; 4 h per day | Sprague Dawley rats | = | [32] |
| 10.5% | 4–21 ED | Sprague Dawley rats | ↓ | [33] |
| 10.5% | 15–21 ED | Sprague Dawley rats | ↓ | [34,35] |
| 10.5% | 11–17.5 ED | BALB/c mice | ↓ | [36] |
| 10.5% | last 15 days of gravidity | guinea pigs | ↓ | [37] |
| 10 ± 1% | 7–21 ED; 3 h per day | Sprague Dawley rats | ↓ | [38] |
| 11% | 15–21 ED | rats | ↓ | [39] |
| 11.5% | 13–20 ED | Sprague Dawley rats | ↓ | [40] |
| 12% | 15–19 ED | Sprague Dawley rats | = | [41] |
| 12% | 14.5–21 ED | CD-1 mice | ↓ | [11] |
| 13% | 6–20 ED | Wistar rats | = | [2,12,42] |
| 13–14% | 6–20 ED | Wistar rats | = | [43] |
| 14% | 6–18 ED | C57BL/J6 mice | = | [44] |
| 15% | 19 ED–delivery; 10 min; 6 times per day | Sprague Dawley rats | = | [45] |
| NA | NA | Jackson Black C-57 mice | = | [46] |
| 280–300 mmHg; 8000 m above sea level | 14 ED–delivery; 2 h per day | C57BL/6 mice | = | [47] |
| PaO2 13 mmHg | 14 days | sheep | = | [48] |
| 3820 m above sea level | 30–120 ED | sheep | = | [49] |
| 4000 m above sea level on first day, 5000 m above sea level on the second to fifth day | 14–18 ED, 8 h per day | rats | ↓ | [50] |
| chronic anaemia | NA | sheep | = | [51] |
| 9000 m above sea level; PaO2 42 mmHg | 14–19 ED, once 4 h | albino rats | ↓ | [52] |
| NA | 105–138 ED | sheep | ↓ | [53] |
ED, embryonic day; NA, non-available; ↓, decreased birth body weight; =, no changes of the birth body weight in comparison with control group.
The most important signalling molecules that respond to changes in oxygen content are hypoxia-inducible factors (HIFs) from the family of heterodimeric transcription factors. The HIF transcription factor is composed of two subunits, α and β. Under normoxia, HIF-1α is rapidly degraded, but if the partial pressure of oxygen decreases, it is stabilized and accumulates in the cells. HIF-1α maintains oxygen homeostasis by regulating the expression of hundreds of genes and interacts with pathways involved in the regulation of the cardiovascular system, such as the chemoreflex, sympathetic drive [54], renin-angiotensin-aldosterone system (RAAS) [55,56], and local vessel wall components such as nitric oxide (NO) [57,58,59] and endothelin-1 [60,61]. Moreover, HIF directly interacts with the circadian clock because HIF-1α controls the expression of several canonical circadian genes and the response to hypoxia is gated by the circadian clock [62]. The relationship between the HIF and clock can be reciprocal, because in several experimental models, hypoxia was found to reduce the amplitude of circadian rhythms or their response to phase shifts [63].
2. Prenatal Hypoxia
Prenatal hypoxia is a common complication in pregnancy, developing from various causes (Figure 1). In gravidity, prenatal hypoxia often occurs as a comorbidity of maternal diseases, such as hypertension, anaemia and respiratory diseases or as a consequence of poor maternal lifestyle, such as smoking, so-called preplacental hypoxia. Prenatal hypoxia can also develop as a consequence of morphological or functional changes in the placenta, such as failed remodelling of spiral arteries or metabolic reprogramming (uteroplacental hypoxia). Disturbed fetoplacental perfusion due to changes in foetal circulation or genetic anomalies in the circulatory system can lead to postplacental hypoxia [64].
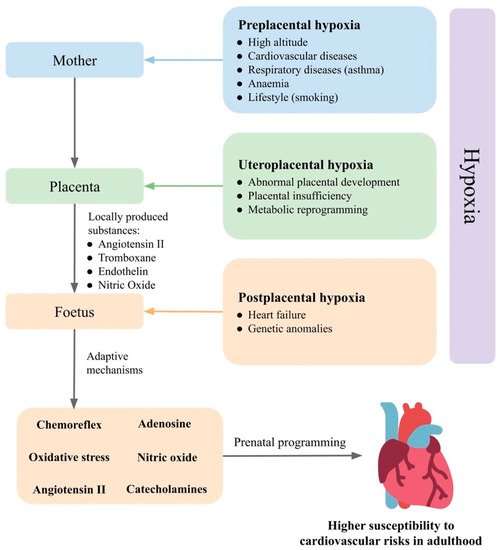
Figure 1. Prenatal hypoxia as result of mother, placenta, and foetus and their effects on cardiovascular regulatory mechanisms in the foetus. Based on the level in which prenatal hypoxia occurs, it can be divided into preplacental hypoxia, uteroplacental hypoxia, and postplacental hypoxia. The foetus is able to compensate for oxygen deficiency and maintain homeostasis by the activation of regulatory mechanisms. The response of the foetus to prenatal hypoxia can also be modulated by the placenta and the mother. Changes in the set points of the cardiovascular regulatory mechanisms in the foetus increase susceptibility to hypertension in adulthood.
2.1. Foetus
Foetal blood is hypoxic compared with maternal blood. The foetal partial pressure of oxygen is reduced to 20–30 mmHg, while in the mother, the arterial partial pressure of oxygen is about 75–100 mmHg at sea level [65,66,67]. Nevertheless, the foetus can cope with up to a 50% (10–15 mmHg) reduction of the partial pressure of oxygen [68]. This has been well-described in sheep used as a common animal model of prenatal hypoxia. In sheep, during normoxia, the maternal partial pressure of oxygen is about 100 mmHg. In comparison, during hypoxia, it can decrease up to 35 mmHg, and the foetus can compensate for this condition. In the case of a sheep foetus, due to hypoxia, the oxygen saturation can decrease from 20 mmHg to 10.5 mmHg in the umbilical artery and from 34 mmHg to 15 mmHg in the umbilical vein [69]. A decrease of the partial pressure of oxygen in the foetus causes femoral vasoconstriction because of local and neurohumoral regulatory mechanisms [70], which can cooperate with the foetal, placental, or maternal regulations.
The adaptive response to hypoxia is a combination of the following five factors:
- (1) Reflex response mediated by the chemoreflex and α1-adrenergic signalling in peripheral vessels [70,71,72]. Hypoxia also directly affects chromaffin cells in the adrenal medulla, thereby stimulating the release of catecholamines. Chromaffin cells of the adrenal medulla have a chemosensitive function until a sympathetic innervation develops between the adrenal glands and the central nervous system [73].
- (2) An endocrine response that is involved in maintaining peripheral vasoconstriction and is activated within about 15 min of the onset of hypoxia, including angiotensin II (Ang II) and other vasoactive substances [71,74,75].
- (3) Local components that respond to the direct effect of hypoxia at the tissue level. The vascular endothelium acts as a hypoxic sensor but also an effector system that releases NO [76], adenosine [77], and endothelin-1 [78], thus affecting the function of vascular smooth muscle.
- (4) Placental vasoactive substances are released from the placenta into the foetus and can thus modulate the response to hypoxia. Such substances include adenosine [79], Ang II, thromboxane, endothelin, NO [80], glucocorticoids and others [81].
- (5) Maternal factors that pass from the mother to the foetus. Environmental hypoxia, during which both the mother and the foetus are hypoxic, causes adaptive changes not only at the level of the foetus but also in the mother. Some of the mother’s adaptation mechanisms may also affect foetal development [81].
Activation of the described mechanisms as a response to prenatal hypoxia ensures sufficient blood flow to vital organs, such as the brain and heart, by peripheral vasoconstriction and reduction in oxygen consumption by the tissues and organs. We further describe the effects of prenatal hypoxia on cardiovascular regulatory mechanisms: the chemoreflex, adenosine, NO, ROS, and RAAS, as well as the effects of prenatal hypoxia on morphological-functional changes in the heart, blood vessels, and kidneys (Table 2).
Table 2. Effects of prenatal hypoxia on the regulatory mechanism of blood pressure.
| Prenatal Hypoxia Type | Animal Model | Hypoxia Outcomes | Ref. | |
|---|---|---|---|---|
| Adenosine | Arterial PaO2 15 mmHg; 1 h | Sheep | Foetal acidosis, mean arterial pressure increase, a transient heart rate decrease | [77] |
| Hypoxia/anoxia; 20 or 60 min |
A1R+/+, A1R+/− and A1R−/− C57BL mice, hippocampal slices, isolated brainstem spinal cord | Reduction in field excitatory postsynaptic potential | [82] | |
| 10% O2; 7.5–10.5 ED |
A1AR+/+ and A1AR-deficient C57BL/6 mice | Growth retardation, less stabilized HIF-1α protein and cardiac gene expression in A1AR−/− embryos | [83] | |
| 10–12% O2; 30 min; 122–128 ED | Western sheep | Cortical blood flow increase, attenuated by a non-selective adenosine receptor antagonist | [84] | |
| NO, ROS | 13% O2; 6–20 ED |
Wistar rats | Foetus: aortic thickening, enhanced nitrotyrosine staining and increased cardiac HSP70 expression. Adult offspring: impaired NO-dependent relaxation, increased myocardial contractility |
[2] |
| 12% O2; for 4, 7, or 10 days; 58–62 ED | Hartley-Duncan guinea pigs | Increased eNOS mRNA in foetal ventricles, not altered K+-channel activation in response to acetylcholine-stimulated coronary dilation | [59] | |
| 40–50% uteroplacental artery ligation; 25 ED | New Zealand white rabbits | Normal left and right ventricular thickness, increased vessel dilatation; HIF-1α, eNOS, p-eNOS, and iNOS induction suggesting increased NO and oxidative stress in the hearts | [85] | |
| 13% O2; most of gestation (prior to day 5) | Wistar rats | Maternal and placental oxidative stress—prevented by maternal treatment with vitamin C | [42] | |
| 13% O2; 6–20 ED | Wistar rats | Increased LF/HF HRV ratio and baroreflex gain—prevented by vitamin C | [86] | |
| Acute: 10% O2; 0.5 h, 127 ± 1 ED; chronic: 10% O2; 105–138 ED | Welsh Mountain sheep | Mitochondria-derived oxidative stress, endothelial dysfunction and hypertension in adult offspring | [53] | |
| 6% O2; 0.5 h | Welsh Mountain sheep | Increased redistribution of blood flow and the glycemic and plasma catecholamine responses | [87] | |
| 14 ± 0.5% O2; 1–19 ED (embryos underwent euthanasia) | Bovans Brown eggs | Cardiac systolic dysfunction, impaired cardiac contractility and relaxability, increased cardiac sympathetic dominance, endothelial dysfunction in peripheral circulations | [88] | |
| Conceived, gestated, born and studied at Putre Research Station (3600 m above sea level) | Sheep (neonates) | Worsened carotid blood flow, vascular responses to potassium, serotonin, methacholine, and melatonin; diminished endothelial response via NO-independent mechanisms in isolated arteries | [89] | |
| 10.5% O2; 15–21 ED | Sprague Dawley rats | Revealed reprogramming of the mitochondrion | [90] | |
| 11% O2; 15–21 ED |
Sprague Dawley rats | Male and female foetuses: increased oxidative stress in placentas; 7-month-old male and female offspring: cardiac diastolic dysfunction; 13-month-old female offspring: reduced vascular sensitivity to methacholine, 13-month-old male offspring: decreased vascular sensitivity to phenylephrine | [91] | |
| 13–14% O2; 6–20 ED |
Wistar rats | Increased α1-adrenergic reactivity of the cardiovascular system, enhanced reactive hyperemia, sympathetic dominance, hypercontractility and diastolic dysfunction in the heart | [92] | |
| 7% O2; 2 h; 50–55 ED; foetal hearts were harvested at the end of hypoxia | Guinea pigs | Decreased heart ATP, lipid peroxides, 4-hydroxynonenal and malondialdehyde; increased apoptotic index, unremarkable foetal heart morphology, normal postpartum neonatal cardiac function and cerebral histology | [93] | |
| Acute: 220–240 mmHg; 10,000 m above sea level; 4–5 min; 18 ED–delivery; chronic: 280–300 mmHg; 8000 m above sea level); 2 h; 14 ED–delivery | C57BL/6 mice | Acute hypoxia: decreased basal O2 consumption rate and intensity of oxidative phosphorylation by the brain mitochondria of newborn, the activation of the respiratory complex II; chronic hypoxia: increased basal O2 consumption rate and oxidative phosphorylation intensity | [47] | |
| RAAS | 10.5% O2; 6–21 ED |
Sprague Dawley rats | Foetal growth restriction, impaired trophoblast invasion and uteroplacental vascular remodeling, increased plasma ET-1 levels, prepro-ET-1 mRNA, ET-1 type A receptor and AT1 receptor in the kidney and placenta | [78] |
| 12% O2; from 14.5 ED |
CD1 mice | Weaning: both sexes: increased susceptibility to salt-induced cardiac fibrosis; male: renal fibrosis by high salt, increased renal renin mRNA; 12 months: both sexes: increased renal renin mRNA expression and concentrations, male: increased AT1a mRNA expression |
[94] | |
| 10.5% O2; 4–21 ED |
Sprague Dawley rats | Increased superoxide production and decreased SOD expression, enhanced NADPH4, but not NADPH1 or NADPH2 in foetal aortas; increased Ang II-mediated vessel contractions in foetal thoracic aortas blocked by losartan | [33] | |
| Acute isocapnic hypoxaemia (foetal PaO2 12–14 mmHg); 1 h; 110/114–124/128 ED | Sheep foetuses | No effects in foetal heart rate, mean arterial pressure, baro- or chemoreflexes, femoral blood flow, femoral vascular resistance or foetal growth | [48] | |
| Reflex | Aortic PaO2 12–15 mmHg without alterations in foetal PaCO2; 1 h; 124 ED | Welsh Mountain sheep foetuses | Transient bradycardia, femoral vasoconstriction and increases in plasma noradrenaline and adrenaline; the NO clamp: persisted bradycardia, greater peripheral vasoconstrictor and catecholaminergic responses—enhanced the chemoreflex sensitivity | [70] |
| PaO2 15 mmHg; 137–144 ED |
Border Leicester Merino cross sheep | Reduced and delayed the IA-type current | [73] | |
| Aortic PaO2 10–11 mmHg without alterations in foetal PaCO2; 1 h; 117–118 ED | Sheep foetuses | Bradycardia, increased arterial blood pressure, femoral vasoconstriction, blood glucose, lactate concentrations, plasma epinephrine and norepinephrine | [95] | |
| Foetal arterial oxygen saturation by 47.3% (uterine blood flow restriction); 118–126 ED | Sheep foetuses | Bradycardia, not in denervated foetuses, followed by a tachycardia; increased foetal heart rate in denervated foetuses; transiently increased foetal blood pressure in intact foetuses and decrease in denervated foetuses; increased cerebral blood flow in both intact and denervated foetuses; decreased carotid vascular resistance in denervated foetuses | [96] | |
| 10% O2; 5–20 ED |
Sprague Dawley rats | Decreased dopamine content in the carotid bodies; until 3 weeks after birth: hyperventilation and disturbed metabolism | [31] | |
| 10% O2; 5–20 ED |
Sprague Dawley rats | Evaluated resting ventilation and ventilatory response; periphery: reduced tyrosine hydroxylase activity within the first postnatal week and enhanced later; central areas: upregulated tyrosine hydroxylase activity within the first postnatal week and downregulated later | [27] |
ED, embryonic day; O2, oxygen; PaO2, partial pressure of O2; PaCO2, partial pressure of carbon dioxide; A1R, adenosine 1 receptor; HSP70, heat shock protein 70; ROS, reactive oxygen species; RAAS, renin-angiotensin-aldosterone system; NO, nitric oxide; HIF, hypoxia-inducible factor; eNOS, endothelial NO synthase; p-eNOS, phospho-eNOS; iNOS, inducible NO synthase; LF/HF, the ratio of low frequency to high frequency; HRV, heart rate variability; ET-1, endothelin-1; AT1, angiotensin II type 1 receptor; SOD, superoxide dismutase; NADPH, nicotinamide adenine dinucleotide phosphate oxidase; Ang II, angiotensin II.
Effects of prenatal hypoxia have been studied in different animal models, including chicken (hatching 21 days), mice (full-term 21 days), rats (full-term 22 days), guinea pigs (full-term 65 days) and sheep (full-term 147 days) [71,97] and each one has its advantages. Sheep have a relatively similar heart size compared with humans, allowing better observation of the changes in foetal cardiovascular parameters in utero. In contrast, mice and rats have smaller hearts than humans; however, they have a rapid reproduction rate, allowing a relatively fast observation of changes in the postnatal period. In addition, the use of mice in a prenatal hypoxia study allows the creation of knockouts to study selected regulatory mechanisms. The chicken embryo develops without direct humoral contacts with the mother, and the development can be easily manipulated [98]. Mice and rats are altricial species, and their development (e.g., nephrogenesis) continues after birth [99], whereas in precocial humans, sheep and guinea pigs, the development is terminated during the prenatal period [100]. The difference among animal models is also related to the time of organogenesis and the critical period of development, which can partially explain some interspecies variation in the foetal responses to prenatal hypoxia.
2.2. Placenta
Maternal and foetal blood do not mix, but their circulations are in close proximity in a newly formed fetomaternal interface, the placenta. This transient organ provides the environment for the exchange of nutrients and gases between the mother and the foetus and protects the foetus from deleterious environmental factors [134]. Oxygen crosses the placental barrier by simple diffusion down its concentration gradient, so the efficiency of its transport depends mostly on uterine blood flow, placental morphology, and placental metabolism [135]. During pregnancy, uteroplacental blood flow increases several times to meet foetal demands and, therefore, structural changes must constantly occur in the placenta [136,137]. If the placenta is exposed to adverse effects, such as hypoxia, its structure and function must change, thus sparing the developing foetus from oxygen deprivation [138]. On the other hand, when placental development is disturbed, the placental oxygen supply might become limited [43,139].
Placental function is linked to its structure. In humans, maternal spiral arteries deliver oxygenated blood into the space between placental chorionic villi, so the villous brush border membrane is washed directly by maternal blood. On the foetal side of the placenta, chorionic villi encompass foetal capillary networks. Maternal blood is thus separated from foetal circulation by several tissue layers [140]. Although some mammals have different numbers of barrier layers, the placental diffusing capacity remains similar among these species [141]. During the first trimester of human pregnancy, maternal spiral arteries are clogged with trophoblast cells derived from the developing embryo, resulting in fetoplacental hypoxia. At this stage, hypoxia is not pathological, on the contrary, it drives the placental and initial foetal development [142]. Meanwhile, clogged maternal arteries undergo physiological remodelling. This is a crucial process to ensure adequate placental perfusion throughout pregnancy, since foetal oxygenation depends strongly on uteroplacental blood flow. During physiological vascular conversion, the endothelial lining and vascular smooth muscle layer are replaced by fibrinoid, leading to vascular lumen enlargement. Remodelled vessels cannot respond to vasoactive substances to the degree they did before the remodelling [143], meaning that maternal blood flows into the intervillous space more continuously and under lower pressure [144]. Such low-resistance flow protects the chorionic villi and provides adequate time for the exchange of nutrients and gases. Moreover, the pressure difference between the intervillous space and foetal capillaries affects the thickness of the villous membrane, thus influencing the placental diffusing capacity [144].
Abnormal placental development with poor spiral artery remodelling can adversely affect placental haemodynamics and placental diffusing capacity [139,145] and lower foetal oxygenation may result from abnormal villous development [146]. Together, the inadequate conversion of spiral arteries along with abnormal villous development can cause placental insufficiency and jeopardize foetal development [145]. The shallow, or even absent, trophoblast invasion of spiral arteries is considered one of the causes of preeclampsia [147]. In preeclampsia, insufficiently remodelled spiral arteries still have a muscle layer [148,149], so they are more reactive to vasoactive substances [80]. In addition, insufficient remodelling can be accompanied by placental atherosis, which is characterized by fibrinoid necrosis of the vessel wall [150] and thrombosis [151], both contributing to uteroplacental ischaemia-reperfusion injury and subsequent oxidative stress [139]. As a result, intermittent placental perfusion is more frequent throughout the pregnancy than is normal [139]. The intermittent perfusion becomes a problem, especially towards the end of a pregnancy, when fetoplacental oxygen consumption is at its peak [148].
Placental vessels lack innervation, and their reactivity mostly results from locally produced substances [80]. Typically, if shear stress is high, placental endothelial cells produce NO, so placental vascular resistance decreases; however, in placental vessels from growth-restricted foetuses, the shear stress-induced vasodilation is impaired, resulting in a parallel increase in placental vascular resistance [136]. The strongest vasoconstrictor produced by preeclamptic placental tissue is probably thromboxane, but other local substances are involved, such as Ang II and endothelin. Placental vasoreactivity is amplified by decreased prostacyclin and prostaglandin E2 production [80]. Finally, the chronic increase in adenosine concentrations in preeclampsia stimulates the production of anti-angiogenic soluble fms-like tyrosine kinase-1, a non-membrane associated splice variant of receptor 1 for vascular endothelial growth factor (VEGF). It binds the angiogenic VEGF, decreasing its free circulating concentrations and reducing vessel growth and placental vasculature [152].
An inappropriate oxygen environment can induce changes in the placental structure, which might be beneficial for improving foetal oxygenation. A placental barrier can become thinner, capillary diameter increases and uteroplacental vascular resistance decreases, so a more efficient diffusion is achieved [138]. These changes are mediated by HIF-1α target genes and their proteins, such as VEGF and erythropoietin [153]. Moreover, hypoxia can stimulate the expression of arginase-2, an enzyme responsible for decreased NO production [136]. Higgins et al. demonstrated in a murine model that there is an oxygen threshold below which the placenta cannot compensate for the lack of oxygen and intrauterine growth restriction occurs. Their experiment showed that an inhalation of 13% O2 during pregnancy led to structural changes in the placenta (reduction of the thickness of the interhaemal membrane, increased labyrinth zone volume, reduced trophoblast volume, increased placental capacity for transport of nutrients and O2 to the foetus), which spared foetal growth. On the other hand, when pregnant dams inhaled 10% O2, the placental barrier became thicker and the exchange surface area was reduced, so the placental diffusing capacity was negatively affected, and foetal growth restriction occurred.
Along with structural changes, placental metabolism is also modified by hypoxia. The hypoxic placenta consumes less oxygen but increases glucose transport and uptake for anaerobic glycolysis. The placenta is susceptible to oxidative stress due to its high metabolic activity; however, with increasing foetal oxygen requirements and increased metabolic activity, the antioxidant capacity of the placenta gradually increases [154]. Such reprogramming can protect the foetus from growth restriction [155], but if the transplacental glucose transport is decreased and placental consumption is still increased, the foetus becomes hypoglycaemic, resulting in growth restriction [153,156]. Two other factors can affect placental metabolism: the stage of pregnancy when hypoxia occurs and maternal food intake [155]. Hypoxia can modulate maternal food intake, therefore, it might be difficult sometimes to distinguish whether the effects are due to a lack of oxygen or a lack of nutrients. The same applies for glucocorticoids; hypoxia can also induce glucocorticoid secretion [157]. A foetus is protected from maternal glucocorticoids by the placental enzyme 11β-hydroxysteroid dehydrogenase (11β-HSD) type 2, converting the biologically active cortisol to the inactive cortisone. Hypoxia reduces the expression of 11β-HSD type 2 [81] and thus allows more glucocorticoids to cross the placental barrier. In contrast, hypoxia does not affect the expression of 11β-HSD type 1, which converts inactive cortisone to active cortisol [158]. Many articles have analysed the effect of antenatal glucocorticoid exposure on the development of the foetal cardiovascular system [159]. Antenatal glucocorticoids have direct and mediated (Ang II, catecholamines) pressor and morphological effects, such as cardiomyocytes maturation as well as growth and differentiation of the smooth muscle and endothelial cells in vessels [160]. Based on these findings, the placenta integrates multiple signals to compensate for the foetal demands. Whether these adaptations are beneficial for the foetus depends not only on the severity of hypoxia but also on other factors that may buffer or amplify the effects of hypoxia.
2.3. Mother
The foetus is entirely dependent on maternal oxygen supply and therefore, maternal hypoxaemia strongly affects the foetus. Maternal hypoxaemia can arise from different aetiologies, which are related to maternal health conditions, such as maternal haematological disorders, chronic pulmonary disease, and heart disease, or various environmental factors. Hypoxia is usually studied by inhaling low oxygen air, which mimics the environment at high altitudes. Millions of people live permanently in high-altitude environments and are expected to be genetically well adapted. Indeed, women living in high altitudes have a more significant increase in uterine perfusion during gestation and because of that, better foetal outcomes [161]. Maternal health conditions can result in an insufficient oxygen supply, even when the mother herself is not hypoxic. In such cases, foetal hypoxia could be a consequence of reduced uteroplacental perfusion or increased fetoplacental metabolism.
Gestational diabetes is one of the most common complications of pregnancy. According to the studied population, its prevalence ranges from 1.7% to 11.6% [162]. During pregnancy, the maternal metabolism changes to ensure optimal foetal development, and a pregnant woman becomes insulin resistant to provide the foetus with a sufficient amount of glucose. When the pancreas of a hyperglycaemic pregnant woman cannot produce enough insulin to maintain glycaemia [163], more glucose passes through the placental barrier to the foetus, which becomes hyperglycaemic. Consequently, foetal hyperinsulinaemia develops with a consequent increase in size and metabolism of the foetus [164]. These changes are reflected in increased uteroplacental and foetal oxygen consumption [165], and if fetoplacental demands exceed the maternal oxygen supply, hypoxia occurs [166]. Moreover, gestational diabetes is associated with a changing “zigzag” pattern of heart rate variability in the foetus [167]. Similar changes were observed during the second stage of parturition when an overstretched or compressed umbilical cord occurred, or during reduced oxygen availability because of uterine contractions [167]. The observed changes suggest that heart rate variability and the “zigzag” profile may be used as an early marker of prenatal hypoxia and not only in pregnancies with gestational diabetes [167].
Haematological disorders. Epidemiological data show that every fifth pregnant woman is anaemic and in developing countries, the prevalence might even reach 75% [168]. During the first trimester, a woman’s blood volume starts to expand, followed by a later increase in red blood cell mass. These pregnancy-induced changes result in physiological anaemia [169]. Since iron is needed for red blood cell formation, its deficiency reduces the capacity of blood to carry oxygen [170]. Interestingly, compensatory changes may lead to paradoxically higher oxygen content in the umbilical cord [171]. Another example of haematological disorders may be an abnormal, rigid sickle shape of erythrocytes, observed in thalassemia [64].
Pulmonary complications. Pregnancy-induced changes may also contribute to the development of obstructive sleep apnoea, whose prevalence reaches up to 26% in late pregnancy because of a higher maternal body mass index [172]. Obstructive sleep apnoea is characterized by episodes of hypopnoea or even apnoea resulting in maternal intermittent hypoxaemia and hypercapnia [173]. Even though such a shortage of oxygen is not necessarily transmitted to the foetus [174], data indicate the association between obstructive sleep apnoea and adverse foetal growth [175]. Among other respiratory disorders, asthma is the most common during pregnancy, with a worldwide prevalence of 2–13% [176]. When airways are obstructed, ventilation becomes uneven, and maternal arterial oxygen saturation decreases [177]. Acute respiratory diseases such as bronchitis, and pneumonia can lead to maternal respiratory failure, represent a risk for the foetus, and are a common pulmonary problem during pregnancy [64].
Cardiac diseases occur in 1% of pregnant women [178]. Maternal cardiac output increases throughout a normal pregnancy until reaches more than 30% of the non-pregnant value [179]. This physiological change, accompanied by a decrease in systemic resistance, is necessary to ensure adequate foetal oxygenation. If a pregnant woman suffers from heart disease, the heart may not adapt to this increased load [180] leading to arrhythmias, heart failure [181], and pulmonary oedema. In such cases, insufficient gas exchange in maternal lungs causes hypoxaemia also in the foetus [180].
Lifestyle. Maternal hypoxia can also develop due to bad lifestyle habits, such as a high-fat diet, smoking, or alcohol consumption. Consumption of a high-fat diet is associated with lower uteroplacental perfusion. Moreover, if a high-fat diet is accompanied by maternal hyperinsulinaemia, it can result in placental dysfunction [182] and the placental oxygen transport it might become limited. Moreover, obese women are more susceptible to developing the above-mentioned diseases, such as gestational diabetes or obstructive sleep apnoea [183], which may further increase the risk of hypoxia. Active smoking causes a rapid increase in maternal pulse and blood pressure. These cardiovascular changes result from the action of serum catecholamines, whose concentration increases within a few minutes after smoking [184]. When the uterine vessels are constricted, uteroplacental blood flow might become temporarily limited, while maternal concentrations of carboxyhaemoglobin also increase, resulting in a lower oxygen supply for the foetus, thus making hypoxia even more pronounced [184]. A recent study showed that passive smoking might also cause foetal hypoxia [185]. Alcohol consumption results in placental oxidative stress and a subsequent decrease in NO availability [186]. Even drinking coffee during pregnancy could potentially affect foetal oxygenation by stimulating the maternal and placental renin-angiotensin system [187] and maternal catecholamine secretion [188].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23052885
This entry is offline, you can click here to edit this entry!