Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
piggyBac (PB), one of three transposons (PB, Sleeping Beauty (SB), and Tol2) found thus far, has been used for efficient transfection of GOI in various mammalian cells in vitro and in vivo.
- piggyBac
- transposon
- non-viral gene delivery
- electroporation
- hydrodynamics
- genetically modified animals
- gene of interest
- long-term gene expression
- chromosomal integration
1. Systemic Gene Delivery via Tail-Vein Injection of PB
The importance of PB-mediated gene transfer in vivo has frequently been tested using tail-vein injection of a solution containing PB components. Typically, for liver-directed gene delivery, transposon and transposase constructs were administered by HGD, one of the most common methods for in vivo gene delivery through high-speed injection of large volumes of DNA solution [1]. This HGD-based transfection of transposons + PB transposase expression plasmid resulted in sustained GOI expression in the liver and kidney of mice [2]. This HGD-based transfection approach was first developed using mice [1], but has been successfully demonstrated in a broad range of animal models, including rat, rabbit, pig, dog, and monkey [3].
2. Useful for Regulated Gene Expression In Vivo
The PB system enables inducible gene expression in desired tissue in vivo, soley based on the ability of PB to induce efficient chromosomal integration of GOI in a host cell. For instance, Sariday et al. [4] employed a tetracycline-regulated TetOn system by delivering two genes with one expressing the tetracycline activator and a second element containing the tetracycline response element driving GOI expression (i.e., luciferase cDNA). They performed HGD-mediated tail-vein co-injection of the two PB transposons and PB transposase expression plasmid. In the absence of induction, mice exhibited long-term GOI expression without a detectable leak of expression beyond 120 days, but a several fold increase in expression was achieved when mice were injected intraperitoneally with doxycycline for inducible expression.
Nakamura et al. [5] generated a novel mouse model for a hepatic disorder. To induce chromosomal integration of the PB transposon, pT-CETD (carrying a CETD unit (loxP-flanked stop cassette, diphtheria toxin-A chain (DT-A) gene, and poly(A) sites) (Figure 1B) in murine hepatocytes, HGD-based tail-vein injection of a TransIT-EE Hydrodynamic Delivery Solution (Takara Bio Inc., Shiga, Japan; hereafter referred to as TransIT-EE) containing pT-CETD + pTrans (PB transposase expresion plasmid) was performed using ICR mice (Figure 1A). Expression of a fluorescent reporter gene was discernible in liver approximately one month after gene delivery, suggesting chromosomal integration of pT-CETD. Thereafter, to induce Cre-mediated excision of floxed sequence in chromosomally integrated pT-CETD, mice, one month after gene delivery of pT-CETD + pTrans, were subjected to tail-vein administration of a plasmid, called pTR/NCre (in which expression of Cre recombinase gene is under the control of a liver-specific promoter; Figure 1B), using TransIT-EE. As a result, these treated mice suffered from liver injury (Figure 1C(b)), probably due to liver-specific expression of a toxic protein DT-A generated from recombined pT-CETD. This experiment suggests that the PB transposon combined with a Cre/loxP system is a useful regulatable tool for manipulating hepatocyte function in vivo in non-Tg animals.
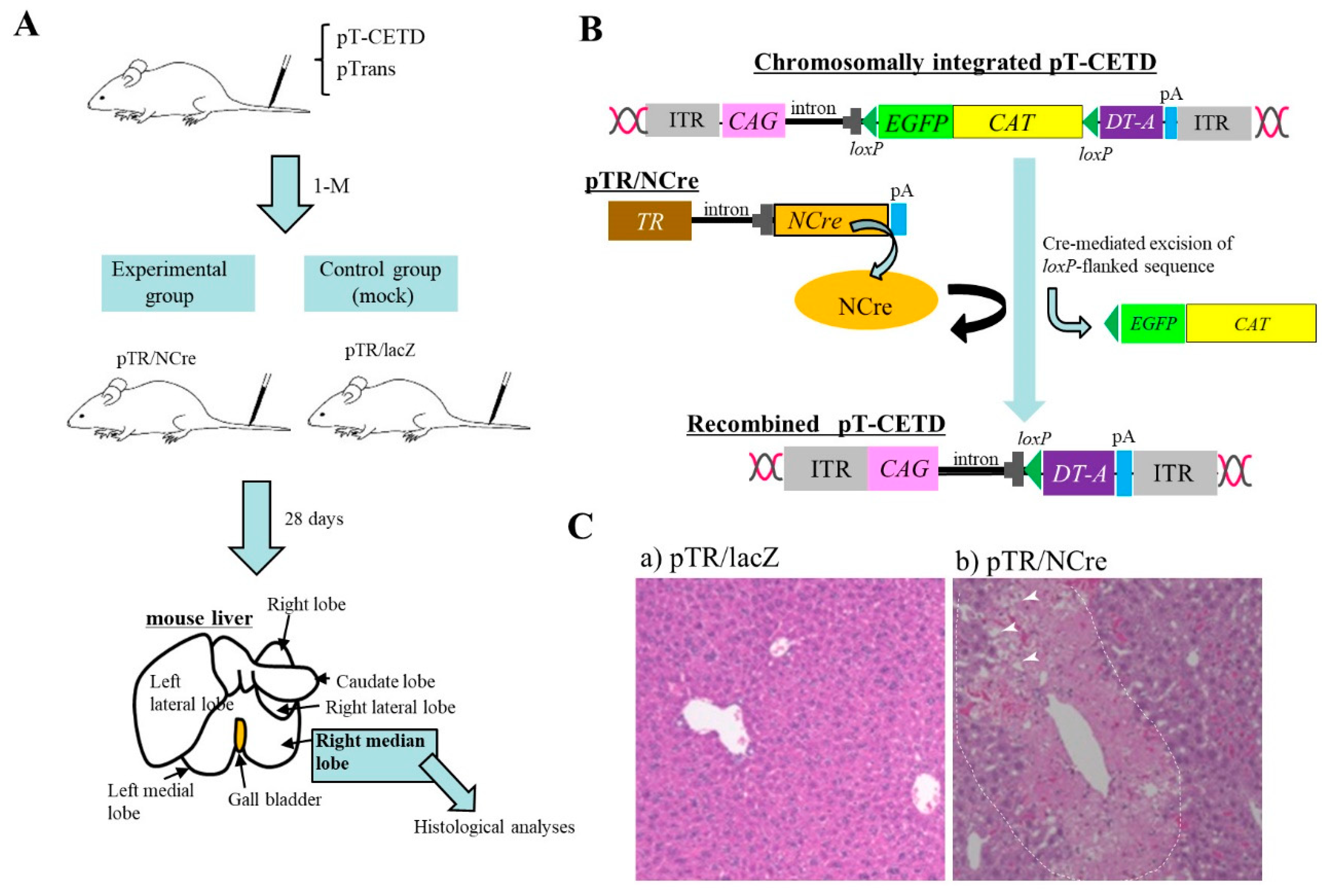
Figure 1. Hydrodynamics-based gene delivery (HGD) and piggyBac (PB) transposon system enable long-term gene expression in murine liver [5]. (A) Schematic representation of experimental outline. At first, PB transposon (pT-CETD) and PB transposase expression plasmid (pTrans) are co-injected into adult ICR male mice via the tail vein by HGD to perform chromosomal integration of CETD component in hepatocytes. One month later, these mice are intravenously injected with pTR/NCre (shown in B) or pTR/lacZ (mock). At 28 days after gene delivery, the right median lobe of liver is dissected for histological analysis. (B) Schematic representation of conditional ablation of murine hepatocytes by Cre/loxP system. When mice are subjected to HGD with a solution containing pT-CETD and pTrans, chromosomal integration of pT-CETD is thought to occur in some hepatocytes (upper panel). Addition of pTR/NCre to these mice via HGD will elicit the generation of recombined pT-CETD, which in turn generates a toxic protein diphtheria toxin-A chain (DT-A) (lower panel). (C) Pathological analysis of pT-CETD-incorporated males assayed 28 days after the second HGD with pTR/lacZ (a) or pTR/NCre (b). Note the presence of focal necrosis (arrowheads in (b); probably caused by conditional expression of DT-A) in an area enclosed by dotted lines. Abbreviations are ITR, inverted terminal repeat; CAG, chicken β-actin-based promoter; EGFP, enhanced green fluorescent protein cDNA; CAT, chloramphenicol acetyltransferase gene; lacZ, gene coding for β-galactosidase; pA, poly(A) sites; TR, transthyretin promoter; NCre, gene coding for Cre with nuclear localization signal.
3. Useful for Transgenic (Tg) Animal Production
Pronuclear microinjection (PI) of nucleic acids (NAs) at the zygote stage has been one of the major ways to produce Tg animals since Gordon et al. [6] first developed it. However, it has always been associated with relatively low Tg efficiency (10–40%) [7][8]. Ding et al. [9] first demonstrated that the PB system is useful for increasing the efficiency of murine transgenesis. They performed PI with a solution containing a transposon carrying a visible marker gene coding for red fluorescent protein (RFP) and a PB transposase expression plasmid. Thus, 35% of pups born (62/184) were transgenic. In comparison, only 10% (10/96) of pups were positive when PI was carried out with the transposon donor alone.
The PB system has also been reported useful for production of Tg domestic animals, such as pigs. In the case of pig zygotes, it is generally hard to visualize pronuclei due to the presence of the lipid droplet layer. Thus, researchers must briefly centrifuge zygotes before microinjection into pronuclei to produce GM pigs [10]. However, this is laborious, as pronuclei are rapidly hindered by the lipid layer within several minutes after centrifugation. Cytoplasmic injection (CI) of DNA appears to be an alternative for the production of Tg animals. Some researchers [11][12] succeeded in creating Tg animals by this procedure, but its success is unstable. This may be due to the ease of delivery of plasmid DNA introduced in cytoplasm of zygotes into nuclei, but rarely integrated into host chromosomes. Li et al. [13] overcame this issue using PB vectors. They performed PI using an all-in-one-type self-inactivating transposon plasmid called pmGENIE-3 (carrying two expression units for transposons and PB transposase) targeting porcine zygotes, and eventually succeeded in producing Tg embryos and piglets. This means that pmGENIE-3 introduced into the cytoplasm of porcine eggs is transferred to nuclei, from which PB transferase mRNA is produced in situ. The resulting mRNA is then transferred to the cytoplasm where the transposase protein is produced. The resultant transposase protein will then bind to ITRs in pmGENIE-3 to generate the transposon/PB transposase complex and cleaved vector backbone. Thereafter, a portion of the former component will be transposed via TTAA present on host chromosomes, as shown in Figure 2.
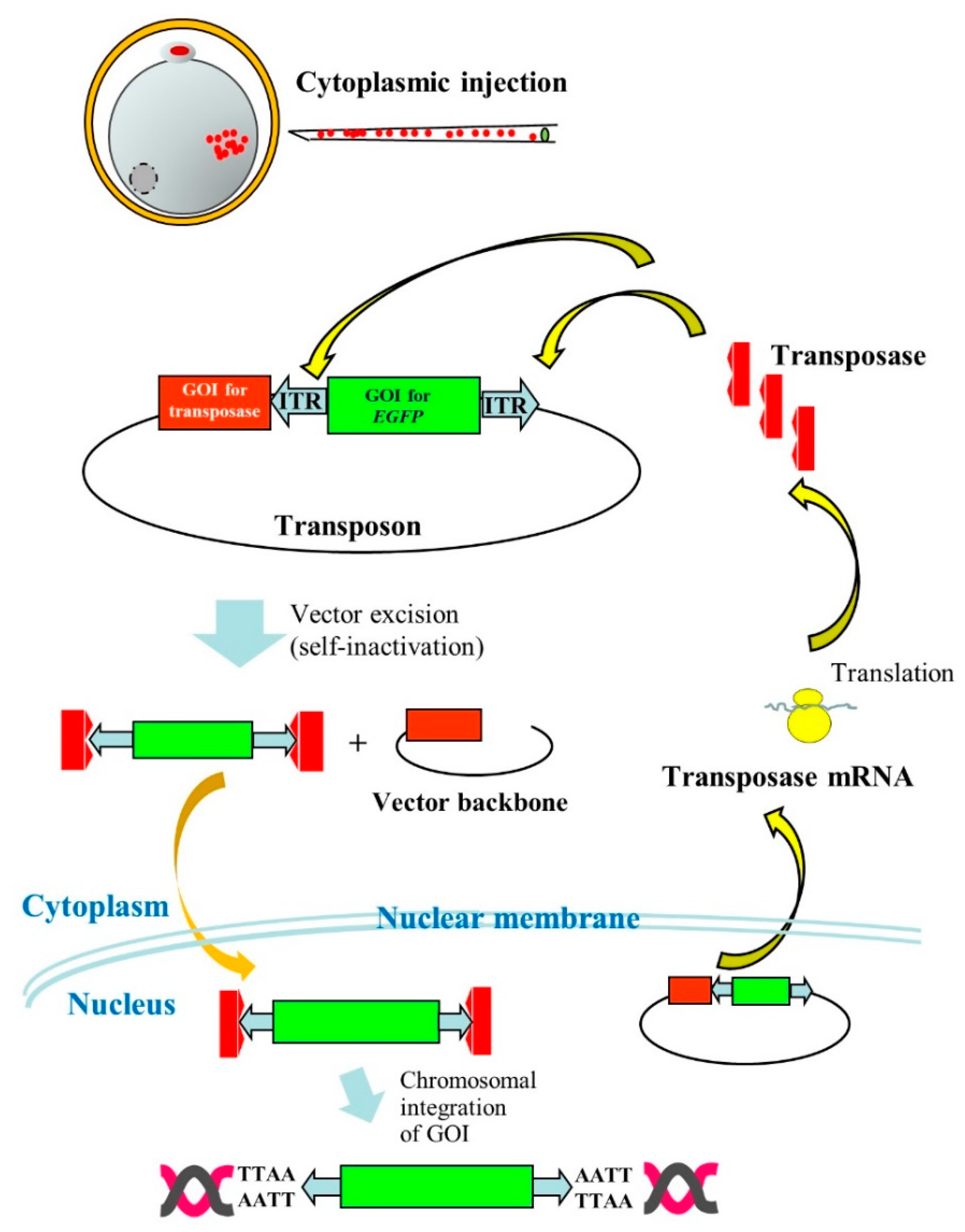
Figure 2. Cytoplasmic injection (CI) of an all-in-one-type piggyBac (PB) vector pmGENIE-3 into porcine parthenogenetically activated oocytes (parthenotes). Following CI of pmGENIE-3, PB transposase mRNA is expressed from the vector and immediately translated into a protein in the cytoplasm. This protein specifically binds to inverted terminal repeat (ITR) present on pmGENIE-3, leading to vector excision. The resulting complex composed of a gene of interest (GOI) for enhanced green fluorescent protein (EGFP) cDNA and PB transposase will be subsequently transferred to the nucleus where chromosomal integration of the GOI will occur.
Tg mice are also produced via the formation of chimeric embryos between normal early mouse embryos (morula or blastocyst) and ES cells that are genetically modified by the PB system [14][15][16]. Rat iPS cells have also been modified by PB to generate Tg rats [17].
Successful production of GM pigs was also demonstrated by somatic cell nuclear transfer (SCNT) of porcine fibroblast cells gene-engineered with the PB system [18].
4. Focal In Vivo PB Gene Delivery
There are several routes for in vivo gene delivery: one is tail-vein injection of DNA using a needle and the other is local administration of DNA into organs/tissue exposed after surgery using a glass micropipette or needle. In some cases, in vivo electroporation (EP) is applied to the injected site to enhance DNA uptake by cells. Local administration of DNA via intramuscular or intradermal injection is also possible without surgery. In these cases, chromosomal insertion of a transgene via a PB-based gene delivery system may be necessary for persistent expression of the transgene. Furthermore, there is an approach for transplanting gene-engineered cells (which have been stably transfected in vitro) to the target organ or tissue for gene therapeutic purposes; this type of experiment is called an “ex vivo experiment.”
4.1. Gene Delivery to Pancreas
The pancreas is an internal organ having important exocrine and endocrine functions in mammals. Diabetes, pancreatitis, and pancreatic cancer are the most common disorders associated with the pancreas. In vivo gene delivery that targets the pancreas is considered as one of the promising approaches for preventing or curing such diseases as well as exploring the biological functions of genes involved in their pathogenesis.
To achieve an efficient gene delivery system targeting pancreatic cells, Sato et al. [19] first employed a non-viral PB-based gene delivery approach coupled with in vivo EP. They injected a solution containing a PB transposon, pT-EGFP (carrying an enhanced green fluorescent protein (EGFP) expression unit), and PB transposase expression plasmid (pTrans) into pancreatic parenchyma of anesthetized adult B6C3F1 female mice under a dissecting microscope with subsequent in vivo EP of the DNA-injected site using tweezer-type electrodes (Figure 3A). Expression of the GOI continued for a minimum of 1.5 months post-gene delivery. The presence of a consensus sequence, TTAA, at the junction between the host chromosomes and transgenes was observed in some samples examined. Sato et al. [19] concluded that such a PB-based gene delivery system could be a useful tool for developing a method to cure diabetes and for exploring other biological applications to assess the function of the pancreas.
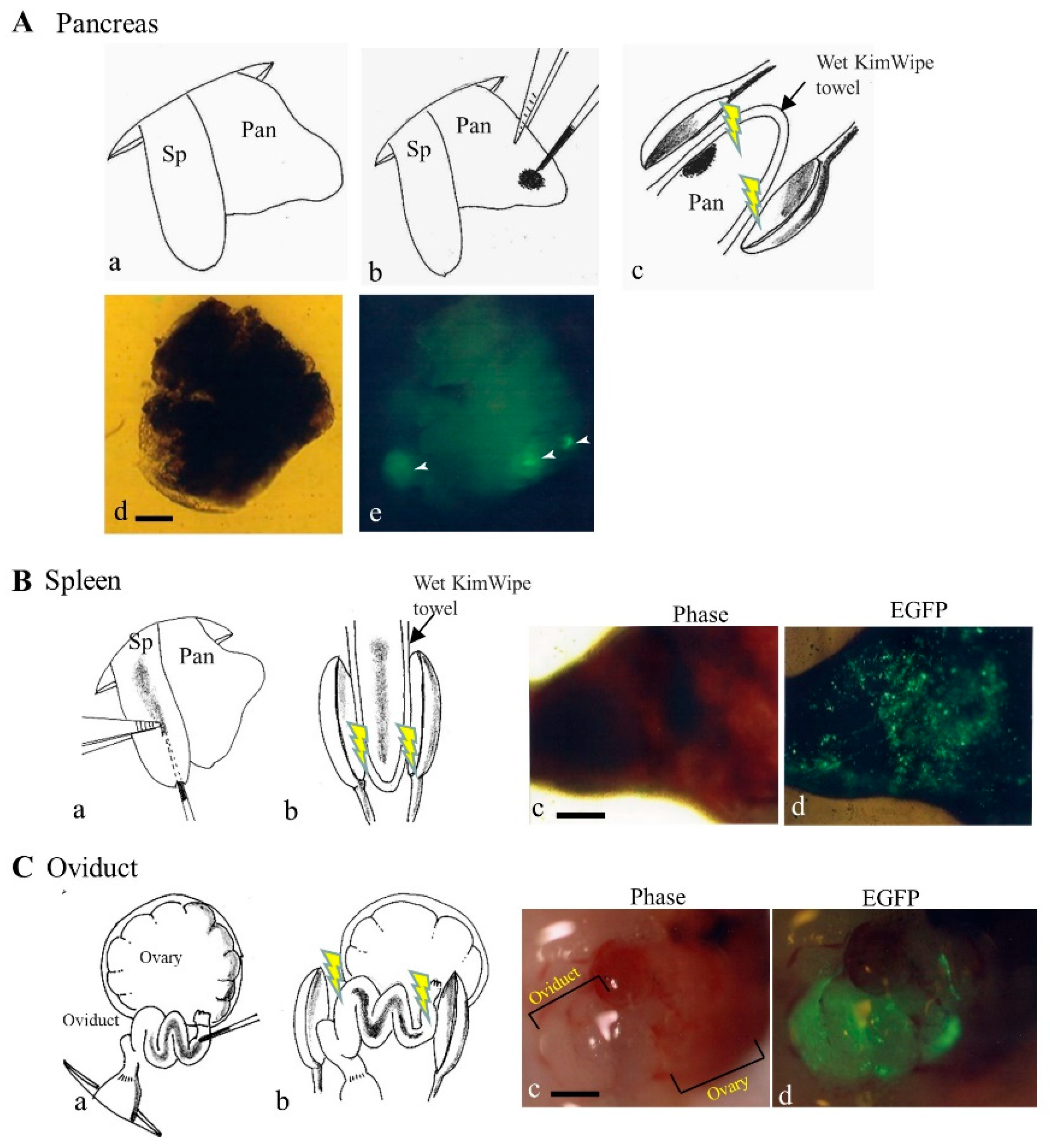
Figure 3. In vivo gene delivery to pancreas (A), spleen (B), and oviduct (C). (A) The gene delivery method to pancreatic parenchyma is illustrated by schematic representation (a–c) and photographs (d,e) [19]. Spleen (Sp) and pancreas (Pan) were exposed after dorsal incision of skin and muscle wall under anesthesia (a). Pancreatic parenchyma was injected with a small volume of solution (1−2 μL) containing enhanced green fluorescent protein (EGFP) expression plasmid DNA and Trypan Blue (b). Thereafter, the injected site of the pancreatic parenchyma was subjected to in vivo electroporation (EP) using two tweezer-type electrodes (c). At ~20 days after gene delivery, dissected pancreas still shows EGFP (arrowheads in (e)). Bar: 1 mm. (B) The gene delivery method to the spleen is illustrated by schematic representation (a,b) and photographs (c,d). Sp was injected with a solution (~20 μL) containing an EGFP expression plasmid DNA and Trypan Blue (a). Thereafter, the injected site was subjected to in vivo EP using two tweezer-type electrodes (b). At 1 day after gene delivery, the dissected spleen showed EGFP-derived fluorescence (c,d). Bar: 1 mm. (C) The gene delivery method to the oviduct is illustrated by schematic representation (a,b) and photographs (c,d). The oviduct was injected with a solution (1−1.5 μL) containing an EGFP expression plasmid DNA and Trypan Blue (a). Thereafter, the entire oviduct was subjected to in vivo EP using two tweezer-type electrodes (b). At 1 day after gene delivery, oviductal epithelium showed EGFP-derived fluorescence (d), but that of untreated intact oviduct did not (c). Bar: 1 mm.
4.2. Gene Delivery to Spleen
The spleen is an organ found in almost all vertebrates and acts primarily as a blood filter. It plays an important role in the immune system, removing old red blood cells while storing white blood cells. Similar to the gene delivery system that targets pancreatic parenchymal cells, the spleen is accessible for gene delivery on the left flank of an anesthetized mouse after laparotomy, because exogenous plasmid DNA can be easily introduced into the spleen under a dissecting microscope.
4.3. Gene Delivery to Oviducts
The oviduct is a part of the mammalian reproductive system, which is required for the fertilization of ovulated oocytes by sperm and their subsequent transport to the uterus, where embryo implantation occurs. It is known that several biologically active factors secreted from oviductal epithelium play an important role in preimplantation embryo development [20].
A direct gene delivery system that targets oviductal epithelial cells appears to be one of the useful approaches to assess detailed biological roles of these cells. An attempt to transfect oviductal epithelium was first made by Relloso and Esponda [21], who injected a solution containing liposomal-encapsulated DNA into the lumen of an oviduct of adult mice and demonstrated that almost all the mice exhibited gene expression in the oviductal mucosa, although only a few number of cells appears to be transfected. Sato [22] used in vivo EP after intra-oviductal instillation of naked plasmid DNA (i.e., an EGFP-expressing vector) to transfect larger numbers of murine oviductal epithelial cells. When the researchers examined this possibility using the same method as that of Sato [22], a large part of the oviductal epithelial cells were found to be successfully (but transiently) transfected, as evidenced by the expression of bright EGFP-derived fluorescence (Figure 3C). It is thus highly expected that gene delivery of the PB transposon system coupled with in vivo EP will enable acquisition of stably transfected oviductal epithelial cells and their long-term expression of the GOIs.
4.4. Gene Delivery to Muscle
PB can be applied to basic research towards the establishment of a therapy model targeting Duchenne muscular dystrophy (DMD), which is a lethal muscle-wasting disease that currently does not have a cure and is caused by a mutated dystrophin gene. In this research area, two approaches, namely cell transplantation- and gene-transfer-based approaches, have been employed. The former case involves ex vivo experiments, whereby isolated muscle progenitor cells are gene-engineered to express the full-length dystrophin gene in vitro and these recombinant cells are then transplanted into the muscle tissue of DMD model animals. The latter case involves direct gene delivery to the injured muscle tissue.
4.5. Gene Delivery to Tail
An all-in-one-type plasmid, called mPB-GLuc-mCherry, which confers simultaneous expression of both mCherry (red fluorescent protein) and GLuc (secretory Gaussia luciferase) together with PB transposase, was injected subcutaneously into the tails of mice followed by in vivo EP locally across the injection site [23]. They observed a GLuc signal six months after gene delivery.
4.6. Gene Delivery to Bladder
The bladder is an organ that cannot be accessed easily by viral vectors due to the presence of a glycosaminoglycan layer covering the urothelium.
4.7. Gene Delivery to Brain
Successful delivery of trophic factors to the brain using stem cell-derived neural progenitors is a promising approach to bypass the blood–brain barrier. Akhtar et al. [24] engineered a PB-based doxycycline-regulated vector, which allows inducible and reversible expression of glial cell line-derived neurotrophic factor (GDNF), a protein known to be effective for protection against neurodegenerative diseases, such as Parkinson’s disease. Nucleofection-based gene delivery of this vector enabled the generation of stably transfected human iPS cell-derived neural progenitors. Transplantation of these stably transfected neural progenitors into an adult non-obese diabetic-severe combined immunodeficiency (NOD-SCID) mouse brain and subsequent addition of doxycycline were found to be effective to induce GDNF expression in vivo. These findings support the usefulness of cell-based therapy using gene-engineered stem cells for possible protection against neurodegenerative diseases.
4.8. Gene Delivery to Kidney
Insulin-like growth factor-1 receptor (IGF-1R) is known to regulate vascular homeostasis and endothelial function. To examine the role of IGF-1R in oxidative stress-induced endothelial dysfunction, Liang et al. [25] constructed a PB-based vector carrying the IGF-1R gene linked to the vascular endothelial (VE)-cadherin promoter. A solution containing the PB transposon and a PB transposase expression plasmid was injected into the renal vein of a mouse kidney, and the mouse was later subjected to unilateral ureteral obstruction (UUO) to induce interstitial fibrosis and inflammatory cell infiltration. Remarkably, a significant reduction in fibrosis (probably due to IGF-1R overexpression in the kidneys) was observed at Day 7 of UUO. The authors [25] concluded that IGF-1R in the endothelium plays a role in maintaining the endothelial barrier function.
4.9. Gene Delivery to Mammary Gland
Mouse mammary glands can regenerate completely by the use of mammary stem cells (MaSCs). To examine the mechanism by which breast cancer develops, Tagaya et al. [26] constructed bacterial artificial chromosome (BAC)-based PB transposons with a vector size of >200 kb to transfect MaSCs via in vitro EP. They transplanted the transfected MaSCs into cleared fat pads of the inguinal mammary glands (from which the endogenous epithelium had been removed) of immune-deficient mice. They observed correct differentiation of the transplanted cells into both basal and luminal cells, as well as milk production after pregnancy. Tagaya et al. [26] also demonstrated that oncogene-induced tumorigenesis is possible when MaSCs transfected with PB transposons, which carry the polyoma-virus middle T antigen gene, are transplanted into mammary glands.
4.10. Gene Delivery to Immune Cells
An emerging approach for treating cancer is the programming of circulating T cells with tumor-recognizing capabilities. In other words, circulating T cells could be gene-engineered through in vitro transfection of genes encoding disease-specific chimeric antigen receptors (CARs), so that the resulting recombinant T cells can combat tumor cells once they are reinfused. For practical use, in vitro production of a large number of tumor-specific T cells appears to be difficult. Recently, an efficient method to quickly program circulating T cells with tumor-recognizing capabilities in mice was reported by Smith et al. [27]. Nanoparticles, co-encapsulated with a transposon plasmid (containing leukemia-targeting CAR genes), and a PB transposase expression plasmid were injected intravenously into mice for allowing preferential uptake by circulating T cells. The nanoparticles used were engineered for selective uptake by lymphocytes through receptor-mediated endocytosis, thereby bringing about long-term disease remission. Consequently, the in situ-engineered T cells exhibited similar activity as that of conventionally engineered T cells generated by ex vivo gene transfer. This approach will be useful as a practical and broadly applicable treatment that can generate anti-tumor immunity “on demand” for oncologists in a variety of settings.
5. In Utero Gene Delivery
In utero EP (IUE) is an effective transfection method for delivering plasmid DNA into neural progenitor cells and neurons of mammalian neocortices of fetal brains [28][29][30][31][32], and fetal tissue including the skin [33], lungs [34], and retinal ganglion cells [35] in vivo. However, introduced plasmid DNA present episomally and is often inactivated or lost after cell division, still remains a problem.
To overcome this, researchers [36][37] demonstrated that IUE, using a PB transposase expression plasmid and transposon plasmids with different promoters that allow for shRNA and bicistronic expression, resulted in stable somatic cellular transgenesis of neurons and glia. These experiments revealed that the PB-based IUE method provides a valuable new tool for tracking and manipulating neural lineages. Recently, Lu et al. [38] applied IUE coupled with PB-mediated somatic mutagenesis to identify potential genes involved in the behavior of cortical neurons in the developing neocortex that often lead to malformations of cortical development (MCDs). IUE was performed at Day 14.5 of the embryonic stage, at the time when cortical neurogenesis is known to be most active. Screening was performed based on the inability of these cells to relocate correctly within the cortex in vivo. In a fetal brain subjected to IUE, extensive random insertional mutations are highly expected. When the insertion sites were assessed using splinkerette-PCR, among the 33 candidate MCD genes identified through this screening, several genes had already been implicated in neural development and disorders. Lu et al. [38] concluded that this approach is able to identify potential mouse genes involved in cortical development and MCD pathogenesis.
6. Application to Gene Therapy
PB can also be efficiently applied for in vivo gene transfer in mice via HGD to correct phenotypes of inherited disease. Liver sinusoidal endothelial cells are a major endogenous source of Factor VIII (FVIII), and the absence of FVIII is known to cause the human congenital bleeding disorder, “hemophilia A”. By correcting hemophilia A-associated phenotypes, Matsui et al. [39] attempted to cure hemophilia A via HGD-based injections of a PB transposon plasmid carrying full-length FVIII cDNA and PB transposase expression plasmid into a hemophilia A mouse model. They observed stable production of circulating FVIII for over 300 days without the development of anti-FVIII antibodies. A similar phenotypic correction of hemophilia A [40] or B [41] was also reported after PB-mediated gene transfer in mouse liver.
As mentioned previously, PB can be applied for therapeutic approaches towards DMD. Cell transplantation-based therapy is always accompanied with heterologous transplantation issues. Recently, the experiments of Iyer et al. [42] suggested the possibility that muscle cell precursors isolated from DMD model mice could be gene-engineered after PB-based transfection with the normal dystrophin gene. If this is realized, autologous transplantation will be possible for curing the dysfunction muscle tissue. In contrast, direct gene-transfer-based therapy has some limitations, as exemplified by transgene silencing issues as well as the difficulty in mediating systemic and sustained dystrophin expression [43].
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics12030277
References
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266.
- Woodard, L.E.; Cheng, J.; Welch, R.C.; Williams, F.M.; Luo, W.; Gewin, L.S.; Wilson, M.H. Kidney-specific transposon-mediated gene transfer in vivo. Sci. Rep. 2017, 7, 44904.
- Tipanee, J.; Chai, Y.C.; VandenDriessche, T.; Chuah, M.K. Preclinical and clinical advances in transposon-based gene therapy. Biosci. Rep. 2017, 37.
- Saridey, S.K.; Liu, L.; Doherty, J.E.; Kaja, A.; Galvan, D.L.; Fletcher, B.S.; Wilson, M.H. PiggyBac transposon-based inducible gene expression in vivo after somatic cell gene transfer. Mol. Ther. 2009, 17, 2115–2120.
- Nakamura, S.; Ishihara, M.; Watanabe, S.; Ando, N.; Ohtsuka, M.; Sato, M. Intravenous delivery of piggyBac transposons as a useful tool for liver-specific gene-switching. Int. J. Mol. Sci. 2018, 19, E3452.
- Gordon, J.W.; Scangos, G.A.; Plotkin, D.J.; Barbosa, J.A.; Ruddle, F.H. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 7380–7384.
- Dunn, D.A.; Pinkert, C.A.; Kooyman, D.L. Foundation Review: Transgenic animals and their impact on the drug discovery industry. Drug Discov. Today 2005, 10, 757–767.
- Houdebine, L.M. Transgenic animal models in biomedical research. Methods Mol. Biol. 2007, 360, 163–202.
- Ding, S.; Wu, X.; Li, G.; Han, M.; Zhuang, Y.; Xu, T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 2005, 122, 473–483.
- Hammer, R.E.; Pursel, V.G.; Rexroad, C.E., Jr.; Wall, R.J.; Bolt, D.J.; Ebert, K.M.; Palmiter, R.D.; Brinster, R.L. Production of transgenic rabbits, sheep and pigs by microinjection. Nature 1985, 315, 680–683.
- Iqbal, K.; Barg-Kues, B.; Broll, S.; Bode, J.; Niemann, H.; Kues, W.A. Cytoplasmic injection of circular plasmids allows targeted expression in mammalian embryos. BioTechniques 2009, 47, 959–968.
- Dunlap-Brown, M.; Butler, S.P.; Velander, W.H.; Gwazdauskas, F.C. Murine embryo development following cytoplasmic injection of linear and condensed DNA. Open J. Anim. Sci. 2012, 2, 23961.
- Li, Z.; Zeng, F.; Meng, F.; Xu, Z.; Zhang, X.; Huang, X.; Tang, F.; Gao, W.; Shi, J.; He, X.; et al. Generation of transgenic pigs by cytoplasmic injection of piggyBac transposase-based pmGENIE-3 plasmids. Biol. Reprod. 2014, 90, 93.
- Wang, W.; Lin, C.; Lu, D.; Ning, Z.; Cox, T.; Melvin, D.; Wang, X.; Bradley, A.; Liu, P. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 2008, 105, 9290–9295.
- Wang, W.; Bradley, A.; Huang, Y. A piggyBac transposon-based genome-wide library of insertionally mutated Blm-deficient murine ES cells. Genome Res. 2009, 19, 667–673.
- Liang, Q.; Kong, J.; Stalker, J.; Bradley, A. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis 2009, 47, 404–408.
- Jiang, M.G.; Li, T.; Feng, C.; Fu, R.; Yuan, Y.; Zhou, Q.; Li, X.; Wan, H.; Wang, L.; Li, W.; et al. Generation of transgenic rats through induced pluripotent stem cells. J. Biol. Chem. 2013, 288, 27150–27158.
- Wu, Z.; Xu, Z.; Zou, X.; Zeng, F.; Shi, J.; Liu, D.; Urschitz, J.; Moisyadi, S.; Li, Z. Pig transgenesis by piggyBac transposition in combination with somatic cell nuclear transfer. Transgenic Res. 2013, 22, 1107–1118.
- Sato, M.; Inada, E.; Saitoh, I.; Nakamura, S.; Watanabe, S. In Vivo piggyBac-based gene delivery towards murine pancreatic parenchyma confers sustained expression of gene of interest. Int. J. Mol. Sci. 2019, 20, 3116.
- Pillai, V.V.; Weber, D.M.; Phinney, B.S.; Selvaraj, V. Profiling of proteins secreted in the bovine oviduct reveals diverse functions of this luminal microenvironment. PLoS ONE 2017, 12, e0188105.
- Relloso, M.; Esponda, P. In-vivo transfection of the female reproductive tract epithelium. Mol. Hum. Reprod. 2000, 6, 1099–1105.
- Sato, M. Intraoviductal introduction of plasmid DNA and subsequent electroporation for efficient in vivo gene transfer to murine oviductal epithelium. Mol. Reprod. Dev. 2005, 71, 321–330.
- Troyanovsky, B.; Bitko, V.; Pastukh, V.; Fouty, B.; Solodushko, V. The Functionality of minimal piggyBac transposons in mammalian cells. Mol. Ther. Nucleic Acids 2016, 5, e369.
- Akhtar, A.A.; Gowing, G.; Kobritz, N.; Savinoff, S.E.; Garcia, L.; Saxon, D.; Cho, N.; Kim, G.; Tom, C.M.; Park, H.; et al. Inducible expression of GDNF in transplanted iPSC-derived neural progenitor cells. Stem Cell Rep. 2018, 10, 1696–1704.
- Liang, M.; Woodard, L.E.; Liang, A.; Luo, J.; Wilson, M.H.; Mitch, W.E.; Cheng, J. Protective role of insulin-like growth factor-1 receptor in endothelial cells against unilateral ureteral obstruction-induced renal fibrosis. Am. J. Pathol. 2015, 185, 1234–1250.
- Tagaya, H.; Ishikawa, K.; Hosokawa, Y.; Kobayashi, S.; Ueoka, Y.; Shimada, M.; Ohashi, Y.; Mikami, H.; Yamamoto, M.; Ihara, T.; et al. A method of producing genetically manipulated mouse mammary gland. Breast Cancer Res. 2019, 21, 1–12.
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820.
- Saito, T.; Nakatsuji, N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 2001, 240, 237–246.
- Tabata, H.; Nakajima, K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: Visualization of neuronal migration in the developing cortex. Neuroscience 2001, 103, 865–872.
- Szczurkowska, J.; Cwetsch, A.W.; dal Maschio, M.; Ghezzi, D.; Ratto, G.M.; Cancedda, L. Targeted in vivo genetic manipulation of the mouse or rat brain by in utero electroporation with a triple-electrode probe. Nat. Protoc. 2016, 11, 399–412.
- Rosin, J.M.; Kurrasch, D.M. In utero electroporation induces cell death and alters embryonic microglia morphology and expression signatures in the developing hypothalamus. J. Neuroinflamm. 2018, 15, 181.
- Huang, C.C.; Carcagno, A. Electroporation of postimplantation mouse embryos in utero. Cold Spring Harb. Protoc. 2018, 2018.
- Sato, M.; Tanigawa, M.; Kikuchi, N. Non-viral gene transfer to surface skin of mid-gestational murine embryos by intraamniotic injection and subsequent electroporation. Mol. Reprod. Dev. 2004, 69, 268–277.
- Henriques-Coelho, T.; Gonzaga, S.; Endo, M.; Zoltick, P.W.; Davey, M.; Leite-Moreira, A.F.; Correia-Pinto, J.; Flake, A.W. Targeted gene transfer to fetal rat lung interstitium by ultrasound-guided intrapulmonary injection. Mol. Ther. 2007, 15, 340–347.
- Garcia-Frigola, C.; Carreres, M.I.; Vegar, C.; Herrera, E. Gene delivery into mouse retinal ganglion cells by in utero electroporation. BMC Dev. Biol. 2007, 7, 103.
- Chen, F.; LoTurco, J. A method for stable transgenesis of radial glia lineage in rat neocortex by piggyBac mediated transposition. J. Neurosci. Methods 2012, 207, 172–180.
- Chen, F.; Maher, B.J.; LoTurco, J.J. piggyBac transposon-mediated cellular transgenesis in mammalian forebrain by in utero electroporation. Cold Spring Harb. Protoc. 2014, 7, 741–749.
- Lu, I.L.; Chen, C.; Tung, C.Y.; Chen, H.H.; Pan, J.P.; Chang, C.H.; Cheng, J.S.; Chen, Y.A.; Wang, C.H.; Huang, C.W.; et al. Identification of genes associated with cortical malformation using a transposon-mediated somatic mutagenesis screen in mice. Nat. Commun. 2018, 9, 2498.
- Matsui, H.; Fujimoto, N.; Sasakawa, N.; Ohinata, Y.; Shima, M.; Yamanaka, S.; Sugimoto, M.; Hotta, A. Delivery of full length factor VIII using a piggyBac transposon vector to correct a mouse model of hemophilia A. PLoS ONE 2014, 9, e104957.
- Staber, J.M.; Pollpeter, M.J.; Arensdorf, A.; Sinn, P.L.; Rutkowski, D.T.; McCray, P.B., Jr. piggyBac-mediated phenotypic correction of factor VIII deficiency. Mol. Ther. Methods Clin. Dev. 2014, 1, 14042.
- Di Matteo, M.; Samara-Kuko, E.; Ward, N.J.; Waddington, S.N.; McVey, J.H.; Chuah, M.K.; VandenDriessche, T. Hyperactive piggyBac transposons for sustained and robust liver-targeted gene therapy. Mol. Ther. 2014, 22, 1614–1624.
- Iyer, P.S.; Mavoungou, L.O.; Ronzoni, F.; Zemla, J.; Schmid-Siegert, E.; Antonini, S.; Neff, L.A.; Dorchies, O.M.; Jaconi, M.; Lekka, M.; et al. Autologous cell therapy approach for Duchenne muscular dystrophy using piggyBac transposons and mesoangioblasts. Mol. Ther. 2018, 26, 1093–1108.
- Puttini, S.; van Zwieten, R.W.; Saugy, D.; Lekka, M.; Hogger, F.; Ley, D.; Kulik, A.J.; Mermod, N. MAR-mediated integration of plasmid vectors for in vivo gene transfer and regulation. BMC Mol. Biol. 2013, 14, 26.
This entry is offline, you can click here to edit this entry!