Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Cell Biology
Systemic sclerosis is a connective tissue disease of unknown origin that is characterized by immune system abnormalities, vascular damage, and extensive fibrosis of the skin and visceral organs. α2-antiplasmin is known to be the main plasmin inhibitor and has various functions such as cell differentiation and cytokine production, as well as the regulation of the maintenance of the immune system, endothelial homeostasis, and extracellular matrix metabolism.
- systemic sclerosis
- α2-antiplasmin
- fibrosis
1. Introduction
Systemic sclerosis (SSc) is an autoimmune rheumatic disease of an unknown origin characterized by immune abnormalities, vascular damage, and fibrosis of the skin and visceral organs [1]. This process usually occurs over many months or years and can lead to organ damage or death. The precise mechanism of SSc progression remains unclear, and there are no therapies to halt the progression of the disease.
The fibrinolytic system dissolves fibrin and maintains vascular homeostasis. The regulators of fibrinolysis contain plasminogen (Plg), a proenzyme which is converted into plasmin by urokinase-type PA (uPA)/uPA receptor (uPAR) or tissue-type plasminogen activator (tPA). The converted-plasmin digests fibrin clots, and fibrin degradation products (FDP) of different molecular weights, including D-dimer, are released into the bloodstream. In contrast, α2-antiplasmin (α2AP) functions as the main inhibitor of plasmin, forming a stable complex plasmin-α2AP (PAP), and results in the inhibition of fibrinolysis [2] (Figure 1). Plasminogen activator inhibitor-1 (PAI-1) binds tPA and uPA and inhibits the generation of plasmin. It has been reported that an uPAR deficiency promotes endothelial dysfunction and fibrosis progression [3,4], and a α2AP deficiency and PAI-1 neutralization attenuate dermal inflammation and fibrosis progression in the bleomycin-induced SSc model mice, and multiple studies suggest that the fibrinolytic factors are associated with the pathology of SSc [5,6,7].
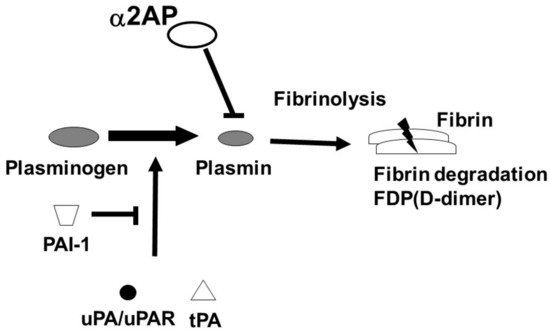
Figure 1. Fibrinolytic system. The fibrinolytic system dissolves fibrin. Plg is converted into plasmin by tPA or uPA/uPAR. The converted-plasmin digests fibrin clots, and FDP (D-dimer) is released into the bloodstream. α2AP functions as the main inhibitor of plasmin and inhibits fibrinolysis. PAI-1 binds and blocks tPA and uPA and inhibits the conversion of Plg to plasmin.
α2AP is a serine protease inhibitor (serpin) that rapidly inactivates plasmin on the fibrin clots or in the circulation [2,8,9]. α2AP has various biological functions independent of plasmin and is associated with thrombosis, angiogenesis, vascular remodeling, fibrosis, brain functions, and bone homeostasis [10,11,12,13,14,15,16,17]. The expression of α2AP is elevated in SSc dermal fibroblasts, and the blockade of α2AP suppresses the progression of pathology in SSc dermal fibroblasts and SSc model mice [18,19]. This review describes the biological functions of α2AP and summarizes the role of α2AP in the progression of SSc.
2. Systemic Sclerosis
SSc is a connective tissue disease of unknown origin characterized by the fibrosis of skin and visceral organs and peripheral circulatory disturbance. The progression of SSc is associated with immune abnormalities (immune cell activation and auto-antibodies production), vascular dysfunction (defective angiogenesis and vasculogenesis, vascular tone alteration, coagulation abnormalities, and endothelial to mesenchymal transition (EndoMT)), and fibrosis (extracellular matrix (ECM) over-production and ECM degradation inhibition) [1]. These abnormalities occur in the various stages of the disease, and these features influence each other and lead to extensive fibrosis and involvement of multiple organs [20,21]. However, the progression of this disease is not completely understood, and there are no therapies to halt the progression of this disease.
3. α2AP
α2AP is a serpin with a molecular weight of 65–70 kd [2] and functions as the main inhibitor of plasmin, a principal component of the fibrinolytic system [2,8,9]. α2AP has been observed in a number of tissues, such as the liver and kidney [22]. Congenital deficiency of α2AP is inherited in an autosomal recessive condition, and individuals with a homozygous α2AP deficiency exhibit severe bleeding symptoms, while heterozygous individuals have mild bleeding tendencies or may be asymptomatic [23,24]. It has been reported that α2AP is associated with pulmonary embolism, ischemic stroke, thrombotic thrombocytopenic purpura (TTP), and arterial thrombosis, and the removal of venous thrombi, wound healing, and fibrosis in several animal studies [6,23,25]. The N-terminal sequence is crosslinked to fibrin by Factor XIIIa (FXIIIa), and the C-terminal region regulates the interaction with plasmin. An antiplasmin-cleaving enzyme (APCE) or fibroblast activation protein (FAP), such as dipeptidyl peptidase 4 (DPP4), causes the cleaving of Met-α2AP to Asn-α2AP (12-amino-acid residue shorter form) [26,27]. It has been reported that Asn-α2AP becomes cross-linked to fibrin approximately 13 times faster than Met-α2AP during clot formation (Figure 2) [28]. In addition, matrix metalloproteinases-3 (MMP-3) inactivates α2AP by cleaving its Pro19-Leu20 peptide bond [29].
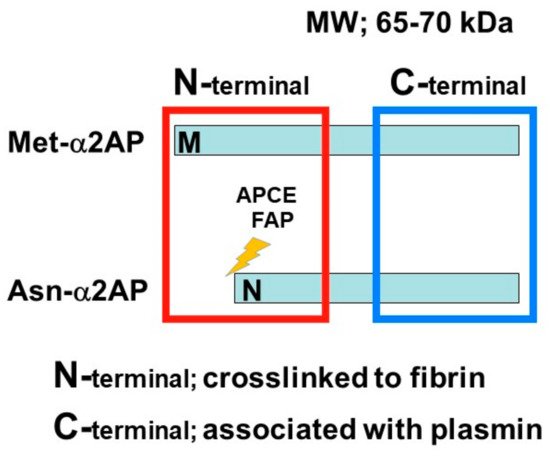
Figure 2. α2AP α2AP is a serpin with a molecular weight of 65–70 kd. The N-terminal sequence is crosslinked to fibrin, and the C-terminal region regulates the interaction with plasmin. Antiplasmin-cleaving enzyme (APCE) or fibroblast activation protein (FAP) causes the cleaving of Met-α2AP to Asn-α2AP (12-amino-acid residue shorter form).
α2AP is most closely related to the noninhibitory serpin pigment epithelium-derived factor (PEDF) [30]. Both α2AP and PEDF have three β-sheets and nine α-helices, and their positions are similar (Figure 3) [31,32]. α2AP and PEDF have very similar structures, and α2AP can bind and activate adipose triglyceride lipase (ATGL), which is known to be a receptor for PEDF [33] and regulates cell signaling, cytokine production, ECM production, cell differentiation, and cell proliferation [5,10,11,34,35]. PEDF has been shown to cause anti-angiogenic effects by inhibiting VEGF signaling [36] and α2AP also inhibits VEGF signaling [19]. α2AP and PEDF may bind the same protein and have similar functions. Furthermore, α2AP contains an arginine-glycine-aspartic acid (RGD) sequence, which is a recognition sequence for integrins [37].
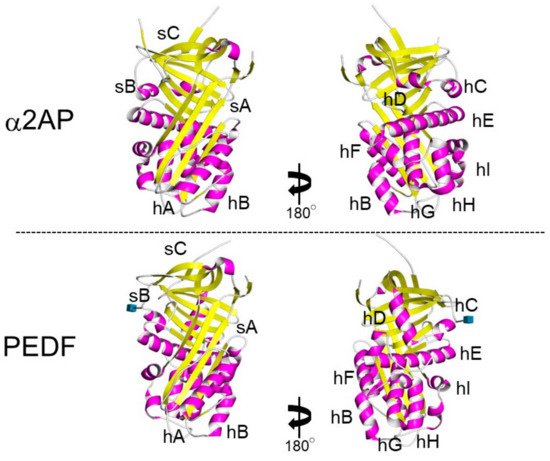
Figure 3. The structure of α2AP and PEDF. The structures of α2AP and PEDF are based on 2R9Y.pdb and 1IMV.pdb, respectively. The three β-sheets are shown in yellow (labeled sA-sC) and the 9 α-helices are shown in pink (labeled hA-hI).
4. α2AP Intracellular Signaling
α2AP can activate multiple intracellular signaling pathways, such as c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase 1/2 (ERK1/2), p38 mitogen-activated protein kinase (MAPK), and src-homology domain-2, containing tyrosine phosphatase 2 (SHP2) [11,13,19,38,39]. In addition, α2AP affects the vascular endothelial growth factor (VEGF) signaling [19], the angiotensin II (AngII) signaling [15], and the advanced glycation end products (AGEs)-induced smad signaling [40]. α2AP and PEDF have a similar structure, and α2AP can activate the adipose triglyceride lipase (ATGL), which is one of the PEDF receptors. PEDF can bind to ATGL, laminin receptor (LR), low-density lipoprotein-related protein 6 (LRP6), and the Notch receptor, and activate various cell signal pathways such as JNK and p38 MAPK [36]. α2AP activates phospholipase A2 (PLA2) through ATGL, which then promotes prostaglandin F2α (PGF2α) synthesis and transforming growth factor-β (TGF-β) production [34]. In addition, α2AP deficiency promotes the expression status of β-catenin, and α2AP attenuates Wnt-3a-induced β-catenin expression and LRP6 activation [12]. Thus, α2AP can activate cell signaling through PEDF-binding proteins, such as ATGL and LRP6, and α2AP may also bind other PEDF-binding proteins and activate various cell signal pathways. On the other hand, α2AP has an RGD sequence at its C-terminus [37], and the RGD sequence affects cell recognition and platelet activation through integrin signaling [41,42].
Plasmin regulates the various signal pathways such as ERK1/2, p38 MAPK, Akt nuclear factor-κB (NF-κB), adenosine monophosphate-activated protein kinase (AMPK), signal transducers, and activators of transcription (STAT) pathways [9,43,44,45]. In addition, plasmin can activate growth factors such as TGF-β, VEGF, basic fibroblast growth factor (bFGF), pro-brain derived neurotrophic factor (proBDNF), insulin-like growth factor-binding protein 5 (IGFBP-5), and hepatocyte growth factor (HGF) [31,46,47]. Furthermore, plasmin can activate MMP-1, MMP-3, MMP-9, and protease-activated receptor-1 (PAR-1), PAR-4, platelets, factors V, VIII, and X [9,48,49,50]. α2AP may regulate various biological functions through plasmin inhibition and α2AP’s self-mediated signaling (Figure 4).
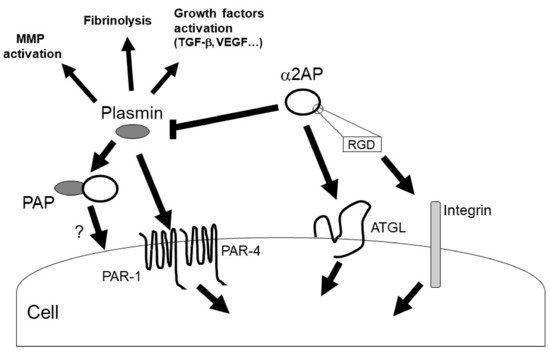
Figure 4. α2AP signaling. α2AP rapidly inactivates plasmin in fibrin clots or in the circulation, resulting in the formation of PAP. α2AP not only inhibits plasmin activity but also activates ATGL and regulates cell signaling. In addition, α2AP contains an RGD sequence and regulates integrin signaling. On the other hand, plasmin has various functions such as fibrinolysis, growth factors and MMPs activation.
5. α2AP Deposition in SSc
The expression of α2AP is elevated in dermal fibroblasts obtained from SSc patients and in the fibrotic tissue of SSc model mice [11,18]. In addition, the levels of PAP in plasma are elevated in patients with SSc [51]. The deposition of α2AP may affect the progression of SSc.
Connective tissue growth factor (CTGF) and interferon-γ (IFN-γ) induce α2AP production through the ERK1/2 and JNK pathways in fibroblasts [11]. In addition, high-mobility group box 1 (HMGB1) induced the production of α2AP through the receptor for advanced glycation end products (RAGE) in fibroblasts [11]. Serum CTGF, IFN-γ, and HMGB1 levels in SSc patients are higher than those in healthy controls [52,53,54]. The blockade of these factors by neutralizing antibodies or inhibitors attenuates dermal fibrosis in bleomycin-induced SSc model mice [54,55]. The increase in these factors may cause the induction of α2AP expression and be associated with the pathogenesis of SSc.
The cleavage of α2AP by MMP-3 inactivates α2AP functions [29,56]. Serum levels of anti-MMP-3 autoantibody and MMP-3 inhibitor, tissue inhibitors of metalloproteinase-1 (TIMP-1) are elevated in SSc patients [57,58]. In addition, the ratio of MMP-3/TIMP-1 is decreased in SSc dermal fibroblasts [56]. The decrease in MMP-3 activity by MMP-3 autoantibody and inhibitors in SSc may suppress α2AP degradation and cause α2AP deposition.
6. α2AP and Immune Abnormalities in SSc
Immune cells such as T cells, B cells, and macrophages have been found in the skin and blood of SSc patients and SSc model mice [59,60,61], and these immune cells have often been observed preceding the fibrotic process [62]. In SSc, B cells cause the production of autoantibodies and the secretion of pro-inflammatory and pro-fibrotic cytokines such as TGF-β, tumor necrosis factor-α (TNF-α), and interleukin-6 (IL-6). B cells also cooperate with fibroblasts, endothelial cells (ECs), and T cells [61], and B cells are associated with EC apoptosis, fibroblast activation, the upregulation of type I collagen synthesis, and regulate the progression of fibrosis and vascular dysfunction in SSc [63,64]. T cells produce various cytokines such as IL-4, IL-5, IL-6, IL-10, and IL-13 [64], and T cell-produced cytokines regulate macrophage activation. In contrast, dermal fibrosis progression is induced in the bleomycin-administrated T and B cell-deficient severe combined immune deficiency (SCID) mice [65,66]. T cells contribute to macrophage activation, but T and B cells may not be essential for the development of fibrosis. Classically (M1) and alternatively (M2) activated macrophages induce pro-fibrotic cytokines such as TGF-β and IL-6 and TIMPs production, fibroblast activation, and collagen production and collagen deposition, and macrophages play a pivotal role in the development of fibrosis in SSc [63,67]. In particular, M2 macrophages are elevated in SSc patients [68]. M2 macrophages are known to be induced by IL-4 and IL-13 [69], and the blockade of IL-4 and IL-13 signaling by IL-4Rα neutralizing antibodies attenuates the progression of fibrosis in SSc model mice [66]. The increase in pro-fibrotic cytokine expression caused by these immune cells is associated with myofibroblast conversion from tissue-resident fibroblast and bone marrow-derived mesenchymal stem cells (MSC), epithelial-to-mesenchymal transition (EMT), and EndoMT. Myofibroblast deposition subsequently promotes excessive ECM production [70,71]. On the other hand, autoantibodies such as anti-MMP-1, anti-MMP-3, and anti-fibrin bound tPA antibodies have been identified in SSc patients, and TIMPs are elevated in SSc [58,72,73,74,75,76], and these factors suppress ECM degradation. Over-production and suppressed degradation of ECM cause fibrosis. Thus, these immune cells are associated with the overproduction of ECM and suppression of ECM degradation and have a major role in the onset of fibrosis in SSc. The correlation between fibrosis progression and the existence of specific autoantibodies such as anti-centromere antibodies and anti-Scl70 antibodies in SSc patients is unknown.
α2AP induces inflammatory cytokine production such as IL-1β and TNF-α [38,39], and α2AP deficiency affects neutrophil recruitment, lymphocyte infiltration, and IgE production [16,77,78]. In addition, PAP causes an increase in IgG and IgM secretion [79]. The blockade of α2AP by α2AP neutralizing antibodies attenuates anti-Scl70 antibody production in SSc model mice [18]. On the other hand, plasmin directly and indirectly regulates cell migration, cell proliferation, cell adhesion, monocyte chemotaxis, macrophage phagocytosis, neutrophil aggregation, monocyte/macrophage infiltration into tissues, and the release of cytokines, growth factors, and other inflammatory mediators [43,80,81,82,83,84,85,86]. In addition, plasmin can activate protease-activated receptor (PAR), platelets, factors V, VIII, and X, and mediate inflammation response [9,48,49]. Furthermore, plasmin effectively cleaves complement factors C3 and C5, thereby releasing the respective chemotactic anaphylatoxin fragments [87], which results in potentiation of TLR4 signaling [88]. α2AP contributes to inflammatory response, immune modulation, antibody production, and plasmin inhibition, and may play an important role as a mediator of inflammation and the immune system in SSc.
This entry is adapted from the peer-reviewed paper 10.3390/life12030396
This entry is offline, you can click here to edit this entry!