Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Lynch-like syndrome (LLS) is defined as colorectal cancer cases with microsatellite instability (MSI) and loss of expression of MLH1, MSH2, MSH6, or PMS2 by immunohistochemistry (IHC) in the absence of a germline mutation in these genes that cannot be explained by BRAF mutation or MLH1 hypermethylation.
- lynch syndrome
- colorectal cancer
1. Introduction
Lynch-like syndrome (LLS) is defined as colorectal cancer cases with microsatellite instability (MSI) and loss of expression of MLH1, MSH2, MSH6, or PMS2 by immunohistochemistry (IHC) in the absence of a germline mutation in these genes that cannot be explained by BRAF mutation or MLH1 hypermethylation [1]. Managing these cases is challenging because the subsequent carcinogenic process is yet to be unveiled. LLS is probably caused by somatic mutations in the mismatch repair (MMR) genes, and, therefore, it is sporadic [2][3]. However, patients with LLS and their relatives have an increased risk of colorectal cancer (CRC), suggesting a possibility of inherited risk. Thus, the most probable scenario is that LLS represents a mixture of sporadic MSI cases, unidentified Lynch syndrome (LS) cases, and possibly other hereditary cases of yet-to-be-determined origin [1][4]. Differentiating between both sporadic and hereditary origin has been a challenge, mainly due to the difficulty in conducting mutational somatic studies of CRC samples.
2. Lynch Syndrome
LS is the most common hereditary cancer syndrome and accounts for approximately 3% of all CRCs [5]. LS is an autosomal dominant disorder caused by germline mutations in MLH1, MSH2, MSH6, and PMS2, as well as deletions in EPCAM. Germline EPCAM deletions result in methylation of the surrounding genomic region, affecting the MSH2 promoter located 18 Kb downstream. As a consequence, MSH2 gene expression is silenced [6]. Constitutional epigenetic silencing of MLH1 [7][8][9][10][11][12][13][14][15][16][17][18][19] and hypermethylation of MSH2 as a consequence of EPCAM deletion [12] have been rarely reported in some families.
LS patients develop multiple tumors, most frequently colorectal and endometrial [20], but also upper gastrointestinal, ovarian, biliary, urinary, brain, non-melanoma skin, and prostate tumors [21]. LS patients are diagnosed at an early age; with a mean age of diagnosis of around 45 years, they develop cancer a mean of 23 years earlier than the general population [22]. Lynch tumors develop faster than sporadic CRC [23]. LS patients have an increased risk of synchronous and metachronous neoplasias. Approximately 7% of LS patients have multiple cancers when diagnosed [23][24]. LS tumors are poorly differentiated, and some present with mucinous features, a medullary growth pattern or showing infiltrating lymphocytes [25][26]. Moreover, their location is predominately in the proximal colon [23][27].
The diagnostic algorithm for LS starts by testing tumors for MSI and/or loss of immunochemical expression of MMR proteins. The Jerusalem guidelines, so-called ‘universal screening’, recommend screening all CRCs and endometrial patients <70 years old for MSI or MMR-D [28]. If MLH1 is lost in IHC, the tumor should then be tested for methylation of the promoter of MLH1 and/or the BRAF V600E mutation to rule out sporadic CIMP tumors. If testing negative, patients are submitted to germline testing, which includes the sequencing and the analysis of deletions and duplications in the appropriate MMR genes. Germline testing results confirm an LS diagnosis [29][30], whereas, if MLH1 is methylated in a tumor, a complementary MLH1 methylation study in blood should be performed to identify constitutional epimutation of MLH1 [31] (Figure 1).
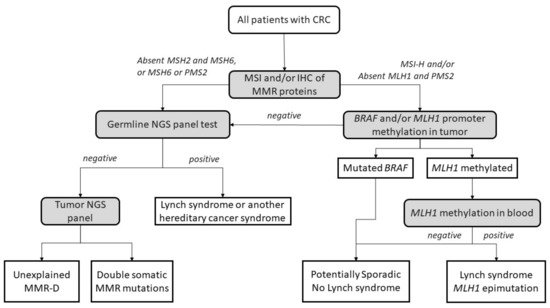
Figure 1. Universal screening strategy for Lynch-syndrome patients. CRC, colorectal cancer; MSI, microsatellite instability; IHC, immunohistochemistry; MMR, mismatch repair; MMR-D, mismatch repair deficiency; NGS, next-generation sequencing. Adapted from Valle et al. [32].
3. Potential Causes of Lynch-like Syndrome
Different plausible causes to explain the origin of LLS tumors have been described. According to the hereditary origin, unknown mechanisms or germline mutations in other genes than those involved in the classical MMR system could mimic the Lynch phenotype with MMR-D. In addition, some LLS cases could be LS with unidentified germline MMR mutations. In contrast, LLS could be due to somatic defects in genes related to tumor onset and progression or due to biallelic alterations in MMR genes outside MLH1 promoter methylation [2][3][33], thus having a sporadic origin. A frequent explanation for LLS cases that should always be ruled out is false-positive IHC/MSI results, which represent approximately 19% of cases in some series [34], and confirmation of MSI and IHC status should be the first step before classifying a case as LLS. Figure 2 describes different potential causes of LLS. It is important to clarify that, if some LLS cases after their molecular analysis can be classified in another category, they will no longer be considered LLS. They will be included in the surveillance program of the new group.
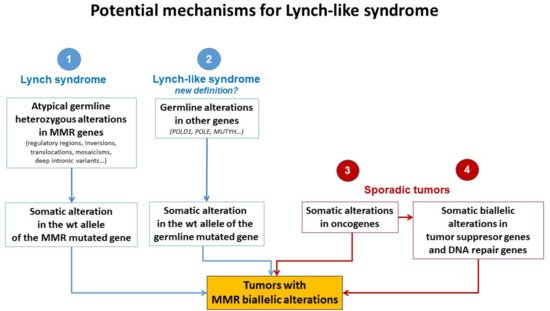
Figure 2. Potential mechanisms for Lynch-like syndrome. MMR, mismatch repair; wt, wild type. Adapted from Pico et al. [35].
3.1. Germline Mutations in Other Genes Affecting the MMR System
The fact that LLS patients are younger at diagnosis than sporadic cases and some of them have a family history of LS-related neoplasias suggests that germline mutations in other genes could also be involved in cancer development in some of these cases (Figure 2). It is important to distinguish whether MMR-D is driving tumor formation or is a secondary event. Germline mutations in MUTYH and POLE have been reported in some patients with MMR-D [14][36][37][38]. Mutations in MUTYH have been previously associated with MUTYH-attenuated polyposis [14][38]. In addition, mutations in MUTYH have been described in MLH1-methylated tumors [14][38]. Approximately 1–3% of LLS cases carry biallelic mutations in MUTYH [14][38]. In addition, mutations in the exonuclease domain of POLE and POLD1 cause a hypermutator phenotype that confers a high predisposition to developing attenuated colorectal polyposis at an early age. POLE and POLD1 mutations may be associated with MMR-D in some cases due to MMR mutations secondary to the hypermutator phenotype [36][37][39][40][41].
On the other hand, Xavier et al., found potentially pathogenic variants in a group of genes involved in the regulation of cellular activity (EXO1, POLD1, RFC1, and RPA1) [42]. EXO1 is related to the union of MLH1 and MSH2, and a mutation in these genes may trigger MMR-D [43]. In addition, RPA1 and POLD1 are associated with harmful effects in tumors with mutations in these genes [44][45]. RFC1 has been described in the development of different malignancies. Huang et al., noted the presence of a variation of this gene in a plasmatic cell tumor [46]. Moreover, somatic mutations in RFC1 were reported in 10.2% of uterine carcinomas and 5.5% of CRCs [47]. This gene also plays an important role in genomic integrity because it is a member of the BRCA1-associated genomic surveillance complex [48]. Golubicki et al., found unknown variants in four genes (POLE, ERCC6, RAD54L, and PALB2) in a group of LLS patients [49]. ERCC6 and PALB2 have been associated with CRC [37][50][51], and the PALB2 variant was previously reported in a suspected case of LS [52].
Next-generation sequencing (NGS) studies have allowed the identification of pathogenic variants that could be candidates for familial CRC with unknown genetic basis. Recently published studies have identified pathogenic variants in genes that maintain DNA integrity resulting in a variety of clinical phenotypes. Germline variants in NTHL1 cause adenomatous polyposis and CRC [53]; MCM9 variants are associated with hereditary mixed polyposis, CRC, and primary ovarian failure [41][54]; and variants in FAN1 cause hereditary CRC by impairing DNA repair [55]. Following this line of inquiry, variants in BUB1 and BUB3 [56], SETD2 [57], WRN [58], BARD1 [58], MCPH1 [58], and REV3L [58] have been found in the germline analysis of LLS cases, linking the mutation of WRN, BARD1, MCPH1, and REV3L for the first time with CRC.
3.2. Hereditary Cases: Unknown Mutations in MMR Genes
In some cases, LLS patients are actually LS patients whose pathogenic variants have not been identified (Figure 2). Current techniques cannot easily identify complex and cryptic mutations. Intronic regions, structural changes such as inversions, and/or copy number variation (CNV) are rarely analyzed genetic changes but may play an important role in unveiling mutations in these patients. For example, the mutation 478 bp upstream of exon 2 in MSH2 creates a canonical splice donor site. The pseudo-exon that is created contains a stop codon that results in a truncated protein [41][59].
Structural changes have been found in some families, such as the inversion of MSH2 exons 1–7 in 10 families in North America [60][61][62] and the inversion of MSH2 exons 2–6 in two families in Australia [63]. Another example of structural genetic changes is the MLH1-LRRFIP2 fusion after a paracentric inversion of chromosome 3 [64] or deletion in that same locus [41][65]. Moreover, Hellen et al., show a retrotranspositional insertion in PMS2 mediated by LINE-1 between exon 7 and 8 [66].
Regulatory regions of MMR genes should also be taken into account. In some cases, variants in the promoter region of MMR have been associated with reduced promoter activity or transcriptional silencing of the allele [57][67]. The accumulation of mutations in the 3′UTR of genes affects mRNA stability and, therefore, protein expression. Germline 3′ UTR mutations in MLH1 have been associated with loss of expression [68]. On the other hand, abnormal regulation of protein expression by miRNA could cause a loss of MMR gene expression. High levels of miRNA-21 downregulate MSH2 and MSH6 and have been found in CRC with loss of MSH2 expression [69]. The same has been seen with MLH1 and miRNA-155 [69]. These examples show the importance of more extensive sequencing methods to detect complex mutations in families of patients with MMR-D and without germline mutations by routine procedures.
Somatic mosaicism could also account for some LLS cases. For instance, Sourrouille et al., described a case of somatic mosaicism in MSH2 after de novo mutation of this gene [33]. Another study described somatic mosaicism in a woman with synchronous gynecological tumors at 44 years old. The MLH1 mutation was only present in 20% of the allele fraction in normal tissue, but her sister and father, who were also affected with LS-related tumors, carried the same mutation [70]. A recent study reported a case of de novo somatic mosaicism in which the MLH1 mutation was detected in the tumor and at a lower level in peripheral blood but not in any other family member [71]. Mosaicism can be detected using highly sensitive NGS with high coverage, and more genetic-driven cases could be correctly identified.
Another important factor to consider is the presence of variants of uncertain significance (VUS) in approximately 30% of cases [72]. Some of them could be pathogenic but cannot be classified due to the absence of clinical, molecular, or functional evidence. Families carrying VUS are managed based on their family history of cancer until further variant classification is available [41].
3.3. Somatic Alteration in Other Cancer Genes or Epigenetic Structures
AT-rich interaction domain 1A (ARID1A) is mutated in a large proportion of tumors [73]. These proteins interact with MSH2, recruiting it to chromatin during DNA replication. Shen et al., demonstrated that impairment of ARID1A contributes to MMR-D [74]. In addition, a somatic exonuclease domain mutation in POLE would be involved in phenocopying defective MMR DNA in 25% of unexplained endometrial cancers with MSI [75].
Local inflammation also promotes genetic and epigenetic alterations in CRC [76] and has been determined to be an important factor in damage to the MMR system [77]. An increase in the concentration of proinflammatory cytokine IL-6 has been demonstrated to alter MMR function. IL6 can activate STAT3, which drives MSH3 out of the nucleus and prevents it from performing its nuclear function [78]. In the same way, high levels of reactive oxygen species (ROS) can induce DNA damage, resulting in MMR-D. Chang et al., showed that non-cytotoxic H2O2 can damage MMR complexes, triggering a reduction in these proteins [79].
Somatic methylation could also explain the MMR-D present in some LLS cases (Figure 2). Many tumor suppressor genes are methylated in sporadic cancers, including RB [80][81], VHL [82], and BRCA1 [83], as well as MLH1 promoter hypermethylation in sporadic CRC caused by the CIMP phenotype [84]. Recently, Buckley et al., reported an association between the methylation of SHPRH and MSI burden [85]. In addition, epimutations in MLH1 and MSH2 have been reported in some families [7][8][9][10][11][12][13][15][16][17][18][19], but other MMR genes can also be targets of somatic methylation [41].
3.4. Somatic Biallelic Alteration in MMR
When comparing tumors with double somatic alterations to LS tumors, no significant histopathological difference was found [86]. Tumor sequencing is the adequate way to evaluate double somatic mutations. Therefore, tumor sequencing should be considered to clarify sporadic versus hereditary causes of unexplained MMR-D [41][87].
Different studies investigated promoter methylation of MMR genes in LLS patients. Methylation of MSH2 was only found in one [87][88] out of 53 LLS patients with loss of expression of MSH2 by IHC studied [57][87][88]. The MSH6 promoter was unmethylated in 108 patients with LLS and MMR-D [57][89][90], and the same happened with the PMS2 promoter in 100 cases with loss of expression of PMS2 or MLH1 who were negative for PMS2 promoter methylation [91]. In summary, based on these studies, somatic promoter hypermethylation of MMR genes does not seem to be the underlying cause of MMR-D in these unexplained tumors.
Therefore, there is a subgroup of LLS that can be explained by double somatic inactivation, and these cases should probably be excluded from the LLS classification due to the probable sporadic origin. However, this approach still has some open questions, because there is no standardized universally accepted technique or protocol for differentiating these cases. Moreover, the biallelic somatic inactivation of MMR genes can also be due to any of the previously described mechanisms, some of them generated by germline genetic alterations. Classifying patients as sporadic or potentially hereditary cases should also be the subject of clinical validation by adequately comparing pedigrees, with long-term follow-up of these families in order to find differences in the incidence of CRC and other LS-related disorders. When some groups advocate for generalization of a somatic study of LLS cases, it is necessary to reach a consensus on how to perform such a study and which cases could be confidently considered as sporadic with no indication for follow-up of patients and relatives. This algorithm has not been clinically validated. Table 1 show a summary of potential causes of LLS.
Table 1. Potential causes of LLS. LLS, Lynch-like syndrome; MMR, mismatch repair.
| Mutations in other Genes Affecting MMR System (Germline) |
Unknown Mutations in MMR Genes (Germline) |
Somatic Mutations in Cancer Genes (Somatic) |
Biallelic Alteration in MMR (Somatic) |
|---|---|---|---|
| MUTYH | Mutation EXON 2 MSH2 | H3K36me3 | Double somatic hit |
| POLE/POLD1 | Inversion EXON 1-7 MSH2 | SETD2 | Somatic mutations in MMR genes |
| EXO1/RFC1/RPA1 | Inversion EXON 2-6 MSH2 | PCNA | Methylation in MMR genes |
| ERCC6/RAD54L/PALB2 | MLH1-LRRFIP2 fusion | ARID1A | |
| PIK3CA | MLH1 3′ UTR mutation | POLE | |
| FAN1/MCM9 | Deep intronic variant in MSH2 | IL-6 and oxidative stress | |
| NTHL1 | miRNA 21 AND miRNA 155 | Methylation in other genes | |
| BUB1/BUB3/WRN/MCPH1/REV3L | Mosaicism | ||
| VUS |
This entry is adapted from the peer-reviewed paper 10.3390/cancers14051115
References
- Rodríguez–Soler, M.; Pérez–Carbonell, L.; Guarinos, C.; Zapater, P.; Castillejo, A.; Barberá, V.M.; Juárez, M.; Bessa, X.; Xicola, R.M.; Clofent, J.; et al. Risk of cancer in cases of suspected lynch syndrome without germline mutation. Gastroenterology 2013, 144, 926–932.
- Mensenkamp, A.R.; Vogelaar, I.P.; van Zelst–Stams, W.A.; Goossens, M.; Ouchene, H.; Hendriks–Cornelissen, S.J.; Kwint, M.P.; Hoogerbrugge, N.; Nagtegaal, I.D.; Ligtenberg, M.J. Somatic mutations in MLH1 and MSH2 are a frequent cause of mismatch-repair deficiency in lynch syndrome-like tumors. Gastroenterology 2014, 146, 643–646.
- Geurts-Giele, W.R.R.; Leenen, C.H.M.; Dubbink, H.J.; Meijssen, I.C.; Post, E.; Sleddens, H.F.B.M.; Kuipers, E.J.; Goverde, A.; Ouweland, A.M.W.V.D.; Van Lier, M.G.F.; et al. Somatic aberrations of mismatch repair genes as a cause of microsatellite-unstable cancers. J. Pathol. 2014, 234, 548–559.
- Picó, M.D.; Sánchez-Heras, A.B.; Castillejo, A.; Giner-Calabuig, M.; Alustiza, M.; Sánchez, A.; Moreira, L.; Pellise, M.; Castells, A.; Llort, G.; et al. Risk of Cancer in Family Members of Patients with Lynch-Like Syndrome. Cancers 2020, 12, 2225.
- Lynch, H.T.; De La Chapelle, A. Genetic susceptibility to non-polyposis colorectal cancer. J. Med. Genet. 1999, 36, 801–818.
- De La Chapelle, A. Genetic predisposition to colorectal cancer. Nat. Rev. Cancer 2004, 4, 769–780.
- Gazzoli, I.; Loda, M.; Garber, J.; Syngal, S.; Kolodner, R.D. A hereditary nonpolyposis colorectal carcinoma case associated with hypermethylation of the MLH1 gene in normal tissue and loss of heterozygosity of the unmethylated allele in the resulting microsatellite instability-high tumor. Cancer Res. 2002, 62, 3925–3928.
- Hitchins, M.; Williams, R.; Cheong, K.; Halani, N.; Lin, V.A.; Packham, D.; Ku, S.; Buckle, A.; Hawkins, N.; Burn, J.; et al. MLH1 Germline Epimutations as a Factor in Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 2005, 129, 1392–1399.
- Hitchins, M.P.; Wong, J.J.; Suthers, G.; Suter, C.M.; Martin, D.I.; Hawkins, N.J.; Ward, R.L. Inheritance of a Cancer-Associated MLH1 Germ-Line Epimutation. N. Engl. J. Med. 2007, 356, 697–705.
- Morak, M.; Schackert, H.K.; Rahner, N.; Betz, B.; Ebert, M.; Walldorf, C.; Royer-Pokora, B.; Schulmann, K.; von Knebel-Doeberitz, M.; Dietmaier, W.; et al. Further evidence for heritability of an epimutation in one of 12 cases with MLH1 promoter methylation in blood cells clinically displaying HNPCC. Eur. J. Hum. Genet. 2008, 16, 804–811.
- Hitchins, M.P.; Ward, R. Constitutional (germline) MLH1 epimutation as an aetiological mechanism for hereditary non-polyposis colorectal cancer. J. Med. Genet. 2009, 46, 793–802.
- Chan, T.L.; Yuen, S.T.; Kong, C.K.; Chan, Y.W.; Chan, A.S.; Ng, W.F.; Tsui, W.Y.; Lo, M.W.; Tam, W.Y.; Li, V.S.; et al. Heritable germline epimutation of MSH2 in a family with hereditary nonpolyposis colorectal cancer. Nat. Genet. 2006, 38, 1178–1183.
- Valle, L.; Carbonell, P.; Fernández, V.; Dotor, A.; Sanz, M.; Benítez, J.; Urioste, M. MLH1 germline epimutations in selected patients with early-onset non-polyposis colorectal cancer. Clin. Genet. 2007, 71, 232–237.
- Castillejo, A.; Vargas-Parra, G.; Castillejo, M.I.; Navarro, M.; Barbera, V.-M.; González, S.; Hernández-Illán, E.; Brunet, J.; Cajal, T.R.Y.; Balmaña, J.; et al. Prevalence of germline MUTYH mutations among Lynch-like syndrome patients. Eur. J. Cancer 2014, 50, 2241–2250.
- Cini, G.; Carnevali, I.; Quaia, M.; Chiaravalli, A.M.; Sala, P.; Giacomini, E.; Maestro, R.; Tibiletti, M.G.; Viel, A. Concomitant mutation and epimutation of the MLH1 gene in a Lynch syndrome family. Carcinogenesis 2015, 36, 452–458.
- Kidambi, T.D.; Blanco, A.; Van Ziffle, J.; Terdiman, J.P. Constitutional MLH1 methylation presenting with colonic polyposis syndrome and not Lynch syndrome. Fam. Cancer 2016, 15, 275–280.
- Hitchins, M.P. Finding the needle in a haystack: Identification of cases of Lynch syndrome with MLH1 epimutation. Fam. Cancer 2016, 15, 413–422.
- Crépin, M.; Dieu, M.-C.; Lejeune, S.; Escande, F.; Boidin, D.; Porchet, N.; Morin, G.; Manouvrier, S.; Mathieu, M.; Buisine, M.-P. Evidence of constitutional MLH1 epimutation associated to transgenerational inheritance of cancer susceptibility. Hum. Mutat. 2011, 33, 180–188.
- Castillejo, A.; Hernández-Illán, E.; Rodriguez-Soler, M.; Pérez-Carbonell, L.; Egoavil, C.; Barberá, V.M.; Castillejo, M.-I.; Guarinos, C.; Martínez-De-Dueñas, E.; Juan, M.-J.; et al. Prevalence ofMLH1constitutional epimutations as a cause of Lynch syndrome in unselected versus selected consecutive series of patients with colorectal cancer. J. Med. Genet. 2015, 52, 498–502.
- Vasen, A.H.F.; Blanco, I.; Aktan-Collan, K.; Gopie, J.P.; Alonso, A.; Aretz, S.; Bernstein, I.; Bertario, L.; Burn, J.; Capella, G.; et al. Revised guidelines for the clinical management of Lynch syndrome (HNPCC): Recommendations by a group of European experts. Gut 2013, 62, 812–823.
- Lynch, H.T.; Boland, C.R.; Rodriguez-Bigas, M.A.; Amos, C.; Lynch, J.P.; Lynch, P.M. Who should be sent for genetic testing in hereditary colorectal cancer syndromes? J. Clin. Oncol. 2007, 25, 3534–3542.
- Hampel, H.; Frankel, W.L.; Martin, E.; Arnold, M.; Khanduja, K.; Kuebler, P.; Nakagawa, H.; Sotamaa, K.; Prior, T.W.; Westman, J.; et al. Screening for the Lynch Syndrome (Hereditary Nonpolyposis Colorectal Cancer). N. Engl. J. Med. 2005, 352, 1851–1860.
- Lynch, H.T.; Snyder, C.L.; Shaw, T.G.; Heinen, C.D.; Hitchins, M.P. Milestones of Lynch syndrome: 1895–2015. Nat. Cancer 2015, 15, 181–194.
- Risio, M.; Reato, G.; Di Celle, P.F.; Fizzotti, M.; Rossini, F.P.; Foà, R. Microsatellite instability is associated with the histological features of the tumor in nonfamilial colorectal cancer. Cancer Res. 1996, 56, 5470–5474.
- Willett, C.G.; Chang, D.T.; Czito, B.G.; Meyer, J.; Wo, J. Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Schwitalle, Y.; Kloor, M.; Eiermann, S.; Linnebacher, M.; Kienle, P.; Knaebel, H.P.; Tariverdian, M.; Benner, A.; von Knebel Doeberitz, M. Immune Response Against Frameshift-Induced Neopeptides in HNPCC Patients and Healthy HNPCC Mutation Carriers. Gastroenterology 2008, 134, 988–997.
- Mas-Moya, J.; Dudley, B.; Brand, R.E.; Thull, D.; Bahary, N.; Nikiforova, M.N.; Pai, R.K. Clinicopathological comparison of colorectal and endometrial carcinomas in patients with Lynch-like syndrome versus patients with Lynch syndrome. Hum. Pathol. 2015, 46, 1616–1625.
- Boland, C.R.; Shike, M. Report from the Jerusalem Workshop on Lynch Syndrome-Hereditary Nonpolyposis Colorectal Cancer. Gastroenterology 2010, 138, 2197.e1.
- Aran, V.; Victorino, A.P.; Thuler, L.C.; Gil Ferreira, C. Colorectal Cancer: Epidemiology, Disease Mechanisms and Interventions to Reduce Onset and Mortality. Clin. Color. Cancer 2016, 15, 195–203.
- Sinicrope, F.A. Lynch Syndrome–Associated Colorectal Cancer. N. Engl. J. Med. 2018, 379, 764–773.
- Hitchins, M.P. Constitutional epimutation as a mechanism for cancer causality and heritability? Nat. Rev. Cancer 2015, 15, 625–634.
- Valle, L.; Vilar, E.; Tavtigian, S.V.; Stoffel, E.M. Genetic predisposition to colorectal cancer: Syndromes, genes, classification of genetic variants and implications for precision medicine. J. Pathol. 2018, 247, 574–588.
- Sourrouille, I.; Coulet, F.; Lefevre, J.H.; Colas, C.; Eyries, M.; Svrcek, M.; Bardier-Dupas, A.; Parc, Y.; Soubrier, F. Somatic mosaicism and double somatic hits can lead to MSI colorectal tumors. Fam. Cancer 2012, 12, 27–33.
- Haraldsdottir, S.; Hampel, H.; Tomsic, J.; Frankel, W.L.; Pearlman, R.; de la Chapelle, A.; Pritchard, C.C. Colon and Endometrial Cancers With Mismatch Repair Deficiency Can Arise From Somatic, Rather Than Germline, Mutations. Gastroenterology 2014, 147, 1308–1316.e1.
- PPicó, M.D.; Castillejo, A.; Murcia, O.; Giner-Calabuig, M.; Alustiza, M.; Sánchez, A.; Moreira, L.; Pellise, M.; Castells, A.; Carrillo-Palau, M.; et al. Clinical and Pathological Characterization of Lynch-Like Syndrome. Clin. Gastroenterol. Hepatol. 2019, 18, 368–374.e1.
- Palles, C.; Cazier, J.B.; Howarth, K.M.; Domingo, E.; Jones, A.M.; Broderick, P.; Kemp, Z.; Spain, S.L.; Guarino, E.; Salguero, I.; et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat. Genet. 2013, 45, 136–143.
- Elsayed, F.A.; Kets, C.M.; Ruano, D.; Akker, B.V.D.; Mensenkamp, A.; Schrumpf, M.; Nielsen, M.; Wijnen, J.T.; Tops, C.M.; Ligtenberg, M.J.; et al. Germline variants in POLE are associated with early onset mismatch repair deficient colorectal cancer. Eur. J. Hum. Genet. 2014, 23, 1080–1084.
- Morak, M.; Heidenreich, B.; Keller, G.; Hampel, H.; Laner, A.; De La Chapelle, A.; Holinski-Feder, E. Biallelic MUTYH mutations can mimic Lynch syndrome. Eur. J. Hum. Genet. 2014, 22, 1334–1337.
- Bellido, F.; Pineda, M.; Aiza, G.; Valdés-Mas, R.; Navarro, M.; Puente, D.A.; Pons, T.; González, S.; Iglesias, S.; Darder, E.; et al. POLE and POLD1 mutations in 529 kindred with familial colorectal cancer and/or polyposis: Review of reported cases and recommendations for genetic testing and surveillance. Genet. Med. 2016, 18, 325–332.
- Spier, I.; Holzapfel, S.; Altmüller, J.; Zhao, B.; Horpaopan, S.; Vogt, S.; Chen, S.; Morak, M.; Raeder, S.; Kayser, K.; et al. Frequency and phenotypic spectrum of germline mutations in POLE and seven other polymerase genes in 266 patients with colorectal adenomas and carcinomas. Int. J. Cancer 2015, 137, 320–331.
- Giner-Calabuig, M. Novel Germline and Somatic Processes in Mismatch Repair Deficient Tumors. Ph.D. Thesis, Universidad de Alicante, Alicante, Spain, 30 November 2020.
- Xavier, A.; Olsen, M.F.; Lavik, L.A.; Johansen, J.; Singh, A.K.; Sjursen, W.; Scott, R.J.; Talseth-Palmer, B.A. Comprehensive mismatch repair gene panel identifies variants in patients with Lynch-like syndrome. Mol. Genet. Genom. Med. 2019, 7, e850.
- Keijzers, G.; Liu, D.; Rasmussen, L.J. Exonuclease 1 and its versatile roles in DNA repair. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 440–451.
- Nicolas, E.; Golemis, E.; Arora, S. POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene 2016, 590, 128–141.
- Wang, Y.; Putnam, C.; Kane, M.F.; Zhang, W.; Edelmann, L.; Russell, R.; Carrión, D.V.; Chin, L.; Kucherlapati, R.; Kolodner, R.D.; et al. Mutation in Rpa1 results in defective DNA double-strand break repair, chromosomal instability and cancer in mice. Nat. Genet. 2005, 37, 750–755.
- Huang, X.; Gao, Y.; He, J.; Cai, J.; Ta, N.; Jiang, H.; Zhu, J.; Zheng, J. The association between RFC1 G80A polymorphism and cancer susceptibility: Evidence from 33 studies. J. Cancer 2016, 7, 144–152.
- The Cancer Genome Atlas Research Network; Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120.
- Wang, Y.; Cortez, D.; Yazdi, P.; Neff, N.; Elledge, S.J.; Qin, J. BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 2000, 14, 927–939.
- Golubicki, M.; Díaz-Gay, M.; Bonjoch, L.; Franch-Expósito, S.; Muñoz, J.; Cuatrecasas, M.; Ocaña, T.; Iseas, S.; Mendez, G.; Carballido, M.; et al. Comprehensive Genomic Characterization of Fifteen Early-Onset Lynch-Like Syndrome Colorectal Cancers. Cancers 2021, 13, 1259.
- Yurgelun, M.B.; Kulke, M.H.; Fuchs, C.S.; Allen, B.A.; Uno, H.; Hornick, J.; Ukaegbu, C.I.; Brais, L.K.; McNamara, P.G.; Mayer, R.J.; et al. Cancer Susceptibility Gene Mutations in Individuals With Colorectal Cancer. J. Clin. Oncol. 2017, 35, 1086–1095.
- Arora, S.; Yan, H.; Cho, I.; Fan, H.-Y.; Luo, B.; Gai, X.; Bodian, D.L.; Vockley, J.G.; Zhou, Y.; Handorf, E.A.; et al. Genetic Variants That Predispose to DNA Double-Strand Breaks in Lymphocytes From a Subset of Patients With Familial Colorectal Carcinomas. Gastroenterology 2015, 149, 1872–1883.e9.
- Yurgelun, M.B.; Allen, B.; Kaldate, R.R.; Bowles, K.R.; Judkins, T.; Kaushik, P.; Roa, B.B.; Wenstrup, R.J.; Hartman, A.-R.; Syngal, S. Identification of a Variety of Mutations in Cancer Predisposition Genes in Patients With Suspected Lynch Syndrome. Gastroenterology 2015, 149, 604–613.e20.
- Weren, R.D.; Ligtenberg, M.J.; Kets, C.M.; De Voer, R.M.; Verwiel, E.T.; Spruijt, L.; van Zelst-Stams, W.A.; Jongmans, M.C.; Gilissen, C.; Hehir-Kwa, J.Y.; et al. A germline homozygous mutation in the base-excision repair gene NTHL1 causes adenomatous polyposis and colorectal cancer. Nat. Genet. 2015, 47, 668–671.
- Goldberg, Y.; Halpern, N.; Hubert, A.; Adler, S.N.; Cohen, S.; Plesser-Duvdevani, M.; Pappo, O.; Shaag, A.; Meiner, V. Mutated MCM9 is associated with predisposition to hereditary mixed polyposis and colorectal cancer in addition to primary ovarian failure. Cancer Genet. 2015, 208, 621–624.
- Seguí, N.; Mina, L.B.; Lázaro, C.; Sanz-Pamplona, R.; Pons, T.; Navarro, M.; Bellido, F.; López-Doriga, A.; Valdés-Mas, R.; Pineda, M.; et al. Germline Mutations in FAN1 Cause Hereditary Colorectal Cancer by Impairing DNA Repair. Gastroenterology 2015, 149, 563–566.
- De Voer, R.M.; van Kessel, A.G.; Weren, R.D.; Ligtenberg, M.J.; Smeets, D.; Fu, L.; Vreede, L.; Kamping, E.J.; Verwiel, E.T.; Hahn, M.M.; et al. Germline mutations in the spindle assembly checkpoint genes BUB1 and BUB3 are risk factors for colorectal cancer. Gastroenterology 2013, 145, 544–547.
- Vargas-Parra, G.M.; González-Acosta, M.; Thompson, B.A.; Gómez, C.; Fernández, A.; Dámaso, E.; Pons, T.; Morak, M.; Del Valle, J.; Iglesias, S.; et al. Elucidating the molecular basis of MSH2-deficient tumors by combined germline and somatic analysis. Int. J. Cancer 2017, 141, 1365–1380.
- Xicola, R.M.; Clark, J.R.; Carroll, T.; Alvikas, J.; Marwaha, P.; Regan, M.R.; Lopez-Giraldez, F.; Choi, J.; Emmadi, R.; Alagiozian-Angelova, V.; et al. Implication of DNA repair genes in Lynch-like syndrome. Fam. Cancer 2019, 18, 331–342.
- Clendenning, M.; Buchanan, D.; Walsh, M.D.; Nagler, B.; Rosty, C.; Thompson, B.; Spurdle, A.; Hopper, J.L.; Jenkins, M.; Young, J. Mutation deep within an intron of MSH2 causes Lynch syndrome. Fam. Cancer 2011, 10, 297–301.
- Mork, M.E.; Rodriguez, A.; Taggart, M.W.; Rodriguez-Bigas, M.A.; Lynch, P.M.; Bannon, S.A.; You, Y.N.; Vilar, E. Identification of MSH2 inversion of exons 1–7 in clinical evaluation of families with suspected Lynch syndrome. Fam. Cancer 2016, 16, 357–361.
- Wagner, A.; van der Klift, H.; Franken, P.; Wijnen, J.; Breukel, C.; Bezrookove, V.; Smits, R.; Kinarsky, Y.; Barrows, A.; Franklin, B.; et al. A 10-Mb paracentric inversion of chromosome arm 2p inactivatesMSH2 and is responsible for hereditary nonpolyposis colorectal cancer in a North-American kindred. Genes Chromosom. Cancer 2002, 35, 49–57.
- Rhees, J.; Arnold, M.; Boland, C.R. Inversion of exons 1–7 of the MSH2 gene is a frequent cause of unexplained Lynch syndrome in one local population. Fam. Cancer 2013, 13, 219–225.
- Liu, Q.; Hesson, L.B.; Nunez, A.C.; Packham, D.; Williams, R.; Ward, R.L.; Sloane, M.A. A cryptic paracentric inversion ofMSH2exons 2–6 causes Lynch syndrome. Carcinogenesis 2015, 37, 10–17.
- Morak, M.; Koehler, U.; Schackert, H.K.; Steinke, V.; Royer-Pokora, B.; Schulmann, K.; Kloor, M.; Höchter, W.; Weingart, J.; Keiling, C.; et al. Biallelic MLH1 SNP cDNA expression or constitutional promoter methylation can hide genomic rearrangements causing Lynch syndrome. J. Med. Genet. 2011, 48, 513–519.
- Meyer, C.; Brieger, A.; Plotz, G.; Weber, N.; Passmann, S.; Dingermann, T.; Zeuzem, S.; Trojan, J.; Marschalek, R. An Interstitial Deletion at 3p21.3 Results in the Genetic Fusion of MLH1 and ITGA9 in a Lynch Syndrome Family. Clin. Cancer Res. 2009, 15, 762–769.
- Van der Klift, H.M.; Tops, C.M.; Hes, F.J.; Devilee, P.; Wijnen, J.T. Insertion of an SVA element, a nonautonomous retrotransposon, inPMS2intron 7 as a novel cause of lynch syndrome. Hum. Mutat. 2012, 33, 1051–1055.
- Liu, Q.; Thompson, B.; Ward, R.; Hesson, L.B.; Sloane, M.A. Understanding the Pathogenicity of Noncoding Mismatch Repair Gene Promoter Variants in Lynch Syndrome. Hum. Mutat. 2016, 37, 417–426.
- Wilding, J.L.; McGowan, S.; Liu, Y.; Bodmer, W.F. Replication error deficient and proficient colorectal cancer gene expression differences caused by 3’UTR polyT sequence deletions. Proc. Natl. Acad. Sci. USA 2010, 107, 21058–21063.
- Valeri, N.; Gasparini, P.; Braconi, C.; Paone, A.; Lovat, F.; Fabbri, M.; Sumani, K.M.; Alder, H.; Amadori, D.; Patel, T.; et al. MicroRNA-21 induces resistance to 5-fluorouracil by down-regulating human DNA MutS homolog 2 (hMSH2). Proc. Natl. Acad. Sci. USA 2010, 107, 21098–21103.
- Pastrello, C.; Fornasarig, M.; Pin, E.; Berto, E.; Pivetta, B.; Viel, A. Somatic mosaicism in a patient with Lynch syndrome. Am. J. Med. Genet. Part A 2009, 149A, 212–215.
- Geurts-Giele, W.R.; Rosenberg, E.H.; Van Rens, A.; Van Leerdam, M.E.; Dinjens, W.N.; Bleeker, F.E. Somatic mosaicism by a de novo MLH1 mutation as a cause of Lynch syndrome. Mol. Genet. Genom. Med. 2019, 7, e00699.
- Thompson, B.A.; Spurdle, A.B.; Plazzer, J.-P.; Greenblatt, M.S.; Akagi, K.; Al-Mulla, F.; Bapat, B.; Bernstein, I.; Capellá, G.; den Dunnen, J.T.; et al. Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database. Nat. Genet. 2014, 46, 107–115.
- Wu, J.N.; Roberts, C.W.M. ARID1A mutations in cancer: Another epigenetic tumor suppressor? Cancer Discov. 2013, 3, 35–43.
- Shen, J.; Ju, Z.; Zhao, W.; Wang, L.; Peng, Y.; Ge, Z.; Nagel, Z.D.; Zou, J.; Wang, C.; Kapoor, P.; et al. ARID1A deficiency promotes mutability and potentiates therapeutic antitumor immunity unleashed by immune checkpoint blockade. Nat. Med. 2018, 24, 556–562.
- Billingsley, C.C.; Cohn, D.E.; Mutch, D.G.; Stephens, J.A.; Suarez, A.A.; Goodfellow, P.J. Polymerase ε (POLE) mutations in endometrial cancer: Clinical outcomes and implications for Lynch syndrome testing. Cancer 2015, 121, 386–394.
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Microbiol. 2014, 12, 661–672.
- Li, S.K.; Martin, A. Mismatch Repair and Colon Cancer: Mechanisms and Therapies Explored. Trends Mol. Med. 2016, 22, 274–289.
- Tseng-Rogenski, S.S.; Hamaya, Y.; Choi, D.Y.; Carethers, J.M. Interleukin 6 Alters Localization of hMSH3, Leading to DNA Mismatch Repair Defects in Colorectal Cancer Cells. Gastroenterology 2015, 148, 579–589.
- Chang, C.L.; Marra, G.; Chauhan, D.P.; Ha, H.T.; Chang, D.K.; Ricciardiello, L.; Randolph, A.; Carethers, J.M.; Boland, C.R. Oxidative stress inactivates the human DNA mismatch repair system. Am. J. Physiol. Cell Physiol. 2002, 283, C148–C154.
- Greger, V.; Passarge, E.; Messmer, E.; Horsthemke, B. Epigenetic changes may contribute to the formation and spontaneous regression of retinoblastoma. Qual. Life Res. 1989, 83, 155–158.
- Sakai, T.; Toguchida, J.; Ohtani, N.; Yandell, D.W.; Rapaport, J.M.; Dryja, T.P. Allele-specific hypermethylation of the retinoblastoma tumor-suppressor gene. Am. J. Hum. Genet. 1991, 48, 880–888.
- Herman, J.G.; Latif, F.; Weng, Y.; Lerman, M.I.; Zbar, B.; Liu, S.; Samid, D.; Duan, D.S.; Gnarra, J.; Linehan, W.M. Silencing of the VHL tumor-suppressor gene by DNA methylation in renal carcinoma. Proc. Natl. Acad. Sci. USA 1994, 91, 9700–9704.
- Dobrovic, A.; Simpfendorfer, D. Methylation of the BRCA1 gene in sporadic breast cancer. Cancer Res. 1997, 57, 3347–3350.
- Kane, M.F.; Loda, M.; Gaida, G.M.; Lipman, J.; Mishra, R.; Goldman, H.; Jessup, J.M.; Kolodner, R. Methylation of the hMLH1 promoter correlates with lack of expression of hMLH1 in sporadic colon tumors and mismatch repair-defective human tumor cell lines. Cancer Res. 1997, 57, 808–811.
- Buckley, A.R.; Ideker, T.; Carter, H.; Harismendy, O.; Schork, N.J. Exome-wide analysis of bi-allelic alterations identifies a Lynch phenotype in The Cancer Genome Atlas. Genome Med. 2018, 10, 69.
- Hemminger, J.A.; Pearlman, R.; Haraldsdottir, S.; Knight, D.; Jonasson, J.G.; Pritchard, C.C.; Hampel, H.; Frankel, W.L. Histology of colorectal adenocarcinoma with double somatic mismatch-repair mutations is indistinguishable from those caused by Lynch syndrome. Hum. Pathol. 2018, 78, 125–130.
- Nagasaka, T.; Rhees, J.; Kloor, M.; Gebert, J.; Naomoto, Y.; Boland, C.R.; Goel, A. Somatic Hypermethylation of MSH2 Is a Frequent Event in Lynch Syndrome Colorectal Cancers. Cancer Res. 2010, 70, 3098–3108.
- Rumilla, K.; Schowalter, K.V.; Lindor, N.M.; Thomas, B.C.; Mensink, K.A.; Gallinger, S.; Holter, S.; Newcomb, P.A.; Potter, J.; Jenkins, M.; et al. Frequency of Deletions of EPCAM (TACSTD1) in MSH2-Associated Lynch Syndrome Cases. J. Mol. Diagn. 2011, 13, 93–99.
- Vymetalkova, V.P.; Slyskova, J.; Korenkova, V.; Bielik, L.; Langerova, L.; Prochazka, P.; Rejhova, A.; Schwarzova, L.; Pardini, B.; Naccarati, A.; et al. Molecular characteristics of mismatch repair genes in sporadic colorectal tumors in Czech patients. BMC Med. Genet. 2014, 15, 17.
- Moura Lima, E.; Ferreira Leal, M.; de Arruda Cardoso Smith, M.; Rodríguez Burbano, R.; Pimentel de Assumpção, P.; Bello, M.J.; Rey, J.A.; Ferreira de Lima, F.; Casartelli, C. DNA mismatch repair gene methylation in gastric cancer in individuals from northern Brazil. Biocell 2008, 32, 237–243.
- Truninger, K.; Menigatti, M.; Luz, J.; Russell, A.; Haider, R.; Gebbers, J.O.; Bannwart, F.; Yurtsever, H.; Neuweiler, J.; Riehle, H.M.; et al. Immunohistochemical analysis reveals high frequency of PMS2 defects in colorectal cancer. Gastroenterology 2005, 128, 1160–1171.
This entry is offline, you can click here to edit this entry!