Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Polymer Science
The synthesis of poly(vinylbenzyl pyridinium salts) fabricated through poly(vinylbenzyl halogene-co-divinylbenzene) quaternization of N-decyloxy-1-(pyridin-3- or -4-yl)ethaneimine was verified by the FTIR spectra of the final quaternized products, in comparison with the spectra of the initial copolymers.
- resins
- Pb(II)
- Cd(II)
- wastewaters
- sorption
1. Introduction
Industrial development, and thus the production of various types of materials and devices, is necessary, but it also involves waste that may cause irreversible changes in the natural environment. Especially, heavy metal pollution in water bodies or groundwater poses a major threat to all living forms [1,2,3]. The presence of heavy metal ions in the environment is mainly caused by the production of wastewater by plants producing paper, fabrics, detergents, and food, but also to a large extent from the chemical, metal refining, petroleum, and petrochemical industries, as well as pharmaceuticals [4]. Some of the heavy metal ions have a positive effect on the functioning of living organisms; for example, zinc is a macroelement that affects the immune system and proper insulin secretion, and also increases sperm production, while copper is a micronutrient involved in the synthesis of collagen and elastin. However, when the permissible concentrations are exceeded, the ions of these metals become toxic. For example, lead tends to accumulate in the blood and soft tissues, which can cause hemoglobin synthesis disorders; damage to the kidneys, liver, lungs, and brain; and even lead to mental retardation and abnormalities in pregnant women [1]. Due to their toxicity and non-biodegradability, effective and selective techniques for the elimination of toxic metals are widely sought [5,6]. The processes for removing heavy metals from wastewater are based on various methods such as chemical precipitation, flotation, ion exchange, and electrochemical deposition, as well as membrane filtration, electrodialysis, and photocatalysis [7,8,9,10]. However, to date, adsorption-based methods have proven to be the most effective and economical in removing toxic metal ions from wastewater [11]. In recent years, various materials of natural origin, agricultural waste, or industrial by-products have been tested as sorbents for removing toxic metal ions; however, only polymer sorbents revealed high adsorption capacity, good regeneration, and good selectivity for certain metal ions. Especially, functional polymers have made it possible to remove a wide range of metals due to the presence of complexing groups in the polymer structure. In particular, resins having carboxyl, phosphonic, or sulfonic functional groups have proved to be of great interest for the sorption of cationic species [12]. However, these resins do not have a significant adsorption capacity, while the regeneration of efficient chelating resins requires a strong acid desorbing agent, such as 4 M HCl in the case of glycidyl methacrylate-based polymer resins [13] or 1 M HNO3 in the case of cross-linked polyzwitterionic acid [14]. In the presented research, the precursors of the pyridinium moieties that were introduced onto the polymer surface are N-decyloxy-1-(pyridnyl)ethanimines with high potential as metal extractants from neutral and acidic aqueous solutions (Fe(III) from HCl [15], Cu(II) and Cu(I) from chloride and sulfate solution [16,17,18], Pb(II) from acidic chloride–nitrate solution [19,20], and Zn(II) from HCl solution [21]). These compounds show exceptionally high metal ion removal efficiency in liquid–liquid and membrane systems [22,23]. For example, using the pseudo-emulsion-based hollow fiber strip dispersion technique (PEHFSD), N-decyloxy-1-(pyridin-3-yl)ethanimine showed much greater potential as the Pd(II) carrier than the commercial extractant (Alamine 308) [23]. The specific properties of those compounds result from the presence in structure of both alkoxyimine and pyridine or pyridinium active sites after quaternization, which through various mechanisms can react with different metals species, e.g., the alkoxyimine group can bind a metal cation through O and N atoms, while the pyridinium cation shows the anion-binding affinity. The high extraction potential of these compounds results not only from the presence of groups capable of coordinating metals, but also from their amphiphilic nature, which enables the interaction in either polar or nonpolar solvent. Moreover, these compounds are also characterized by high stability, which guarantees their long-term use even after contact with strong acidic solutions. Therefore, the functionalization with this type of compounds should provide not only specific sorption properties but also stability, both chemical and thermal.
2. Structural and Thermal Characterization of Resins
The synthesis of VBC-D3EI, VBC-D4EI, VBBr-D3EI, and VBBr-D4EI resins through VBC and VBBr quaternization of N-decyloxy-1-(pyridin-3- or -4-yl)ethaneimine was verified by the FTIR spectra of the final quaternized products, in comparison with the spectra of the initial copolymers. The FT-IR spectra revealed the appearance of a new strong band assigned to C=N vibrations of the pyridinium nitrogen. In the case of VBC-D3EI and VBC-D4EI, the bands were observed at 1631 and 1639 cm−1, respectively, while in the case of VBBr-D3EI and VBBr-D4EI, they were observed at 1623 and 1643 cm−1, respectively. This band was much more intense when the imine substituent was located at position 4 of the pyridine ring. An increase in intensity was also observed at 1022 cm−1, which also confirmed the presence of pyridinium moieties in the structure of the polymer. The imine C=N vibrations were also observed on each spectrum; however, the band was much less intense and was observed at 1608 or 1610 cm−1 for the D3EI and D4EI derivatives, respectively. Regardless of location of the imine substituent in the pyridine ring and structure of the polymer matrix, the spectra also revealed an increase in intensity of peaks at 1508–1510 and 1442–1448 cm−1, which were attributed to C=C vibrations, and the appearance of new peaks assigned to C=N-O bonds (at 1155 cm−1 (VBC-D4EI), 1161 cm−1 (VBC-D3EI), 1168 cm−1 (VBBr-D4EI), and at 1157 cm−1 (VBBr-D3EI). Moreover, similar to the unmodified VBC, the spectra of VBC-D3EI and VBC-D4EI also indicated the presence of the peaks attributed to stretching vibrations of the chloromethylene group (at 1261 cm−1 and at 802 cm−1). In the case of VBBr, VBBr-D3EI, and VBBr-D4EI, peaks observed at 1071 cm−1 and 707 cm−1 were attributed to stretching vibrations –CH2Br and C–Br.
The CP-MS 13C-NMR spectrum of the fabricated resins showed peaks at chemical shifts of 14.9, 26.7, 39.4, and 45.9 ppm, due to aliphatic CH3, CH2, and CH groups. In the case of VBC-D3EI and VBC-D4EI, carbon atoms of the CH2Cl appeared at 64.1 and 64.2 ppm, respectively. Moreover, after modification with D3EI, the signals assigned to aromatic rings were observed at 128.4, 145.8 (increased in intensity), and 152.3 ppm, and the imine C=N appeared at 120.9 ppm. After modification with D4EI, the signals assigned to the aromatic ring observed for VBC at 128.4 ppm increased, reaching a maximum at 122.1 ppm, and new signals appeared at 144.1 and 153.4 ppm. The imine C=N was hidden by pyridine CH atoms. The CP-MS 13C-NMR spectrum of VBBr after quaternization of D3EI and D4EI resulted in chemical shifts of 14.8, 22.6, 29.5, 39.4, and 45.9 ppm, which were assigned to aliphatic CH3, CH2, and CH groups, and signals at 65.3 and at 65.1 ppm corresponding to CH2Br attached to the pyridine nitrogen of D3EI and D4EI, respectively. The pyridine C and CH atoms in VBBr-D3EI were indicated at 127.2, 135.5, and 145.4 ppm, while in VBBr-D4EI they were indicated at 123.7, 144.3, and at 153.1 ppm. The imine C=N was observed at 122.1 ppm.
Based on elemental analyses, the degree of the quaternization and the amounts of the 3- or 4-[1-(decyloxyimine)ethyl]pyridinium groups incorporated in the polymer matrix were calculated. The results are presented in Table 2 and reveal that the involvement of the functional groups in the polymer matrix depend on the structure of the pyridinium moiety and type of counter anion. More effective functionalization was observed for moieties bearing the imine substituent at the 4-position of the pyridine ring than in the case of the 3-position, and quaternization with the chloromethylene moieties occurred more readily than with the bromethylene. The counter-anion had a significant influence on the quaternization reaction. Metzger [32] pointed out that an increase in the basicity of the leaving group shifted the transition state of the quaternization to the products; thus, the more labile group is bromide. It can be assumed that the location of the substituent in the 3-position and Br− with ion radii 0.196 caused a spatial hindrance, limiting the concentration of functional groups in the polymer matrix.
Table 2. Results of elemental, textural, and wettability analysis of the fabricated resins.
| Sample | Degree of Quaternization % |
Cp1 mmol/g |
Pore Size nm |
Specific Surface Area m2/g |
Contact Angle Degree |
|---|---|---|---|---|---|
| VBC | - | 5.0 | - | - | - |
| VBC-D3EI | 90.1 | 2.1 | 6.6 | 33.3 | 105.4 |
| VBC-D4EI | 80.9 | 2.2 | 5.8 | 39.1 | 110.7 |
| VBBr | - | 4.7 | - | - | - |
| VBBr-D3EI | 84.2 | 1.8 | 7.8 | 28.4 | 123.2 |
| VBBr-D4EI | 75.9 | 1.9 | 6.2 | 35.7 | 125.3 |
Cp1, the concentration of pyridinium group.
Full characterization of the functionalized surface required measuring the depth profiles of chemical composition (Figure 2). The C 1s spectra were resolved into four components by peak fitting. The first line centered at 284.8 eV arose from aliphatic and aromatic carbon bonds, the second line lying at 286.5 eV indicated the presence of C-NH bonds, and the third line centered at 289.0 eV indicated the presence of the imine N–C–O bonds [33]. The line at approx. 292 eV was identified as the π → π* shake-up line. This state resulted from 1s photoelectrons emitted from carbon atoms that excited an aromatic ring before leaving the sample surface and is indicative of the presence of aromatic C=C and C=N structures (sp2) [34]. The occurrence of the shake-up peak confirmed the presence of pyridine functionalities on the surface. During sputtering, the C–NH and N–C–O bond components decreased, whereas C–C and C=C increased. The spectra collected in the N 1s region show a single peak at 401.0 eV attributed to pyridine [35,36] and imine nitrogens [37], but for these bands, the changes were not observed during sputtering.
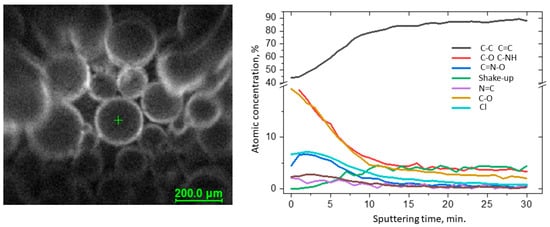
Figure 2. Results of the XPS depth profiling study of VBC-D4EI.
The specific surface area and pore structure of the fabricated resins were also investigated through low-temperature nitrogen sorption. Results showed that the specific surface area varied between 28.4 and 39.1 m2/g and strongly depended on the amount of functionalities incorporated onto the surface of the resins (Table 2). Regardless of the tested resins, the N2 adsorption curves were a type-IV according to the IUPAC classification, indicating the mesoporous nature of the sorbent. Moreover, for all fabricated products, the H3 hysteresis loop was observed, indicating the presence of narrow slit-like pores, as well as particles with internal voids of irregular shape and broad size distribution.
The surface morphologies of VBC and VBBr series resin particles were observed by SEM, and representative images are shown on Figure 3. It could be observed that the surface of all analyzed materials was homogeneous and smooth. It had rare defects and a small number of pores with nanometer diameters. There were no significant differences between the individual materials. Moreover, as in the case of the unfunctionalized polymer matrix, it was observed that more than 90 percent of the imaged grains were in the size range of 175–300 μm (diameter), and the shape of the grains was perfectly spherical, and there was an occasional defect that looked similar to a dent from an adjacent ball.
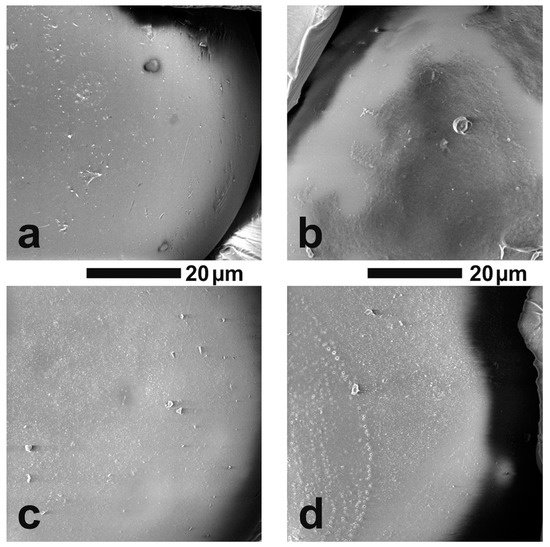
Figure 3. The grain surface of the tested resins at a magnification of 5000×: (a) VBC-D3EI, (b) VBC-D4EI, (c) VBBr-D3EI, (d) VBBr-D4EI.
The measurement of the contact angle provides information on the wettability of water on the resin surface. The results obtained show that the carried-out functionalization of VBC and VBBr with D3EI and D4EI caused poor surface wetting (θ > 90°) (Table 2). A correlation between the structure of functional groups (the substituents’ location and type of the counter anion) and the hydrophilicity of the sorbents was also observed. It was shown that the functionalization with 3-substituted pyridine yielded lower contact angle values than that observed for 4-analogue. Similarly, the presence of Br− as the counter anion made the surface more hydrophobic than in the presence of Cl−.
The thermal properties of the synthesized sorbents were investigated using thermogravimetric analysis (TGA) and differential scanning calorimetry (DSC) methods. In Figure 4, TGA and DTG curves of two types of the studied copolymers VBBr and VBC, as well as products of their modification with two types of functionalities (D3EI and D4EI), are presented. As can be seen, their thermal degradation was a multistep process. The investigated copolymers had three stages of thermal decomposition, while sorbents, after modification, had more decomposition stages, which could be associated with grafted structures resulting from the modification. The first one, which occurred below 100 °C, is related to solvent evaporation. These residual amounts of solvent could be absorbed by the polymer grains during their synthesis and purification. The next two stages are the decomposition of the copolymer: the pendant groups, and then the crosslinked network. The beginning of the thermal decomposition of the copolymers was determined as the temperature of the onset of weight loss and is presented in Table 3. Generally, unmodified sorbents have higher thermal stability (above 300 °C) than modified sorbents (below 250 °C). Comparison with the literature data [25] showed that the obtained sorbents had a higher thermal resistance than previously obtained by us modified VBC and VBBr copolymers with substituents K4.10, K3.10, Ox3.10, and Ox4.10, as well as the commercial Lewatit TP 207 [38] or Amberlyst 15 [39] resin. Additionally, the VBC copolymer was characterized by higher thermal stability (Tonset = 382 °C) than VBBr copolymer (Tonset = 336 °C), by about 50 °C. Despite these differences in the thermal resistance of unmodified copolymers, the introduction of substituents led to materials with similar resistance on thermal degradation. Thus, the thermal resistance was significantly influenced by the substituents introduced into the copolymers during the modification. However, after modification, VBBr-based sorbents had slightly greater thermal stability than those obtained on the basis of VBC. No phase transitions, related to, e.g., glass transition temperature, were observed in the DSC thermograms. This may be due to the highly cross-linked structure of the copolymers. On the other hand, the residual solvent evaporation transition was observed. The heat of this transition was calculated from the thermograms, as well as the temperature at the maximum of the peak of this transition, and the results are presented in Table 3. The heat of evaporation of the solvent in the case of VBBr copolymers increased after modification of their structure, while it decreased in the case of VBC copolymers. This characteristic may be related to the sorption capacity of the synthesized materials. Thus, it may decide on their use as metals ions sorbents.
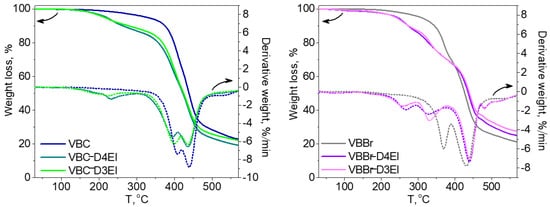
Figure 4. TG and DTG curves of unmodified and modified copolymers: (a) VBC and (b) VBBr with D3EI and D4EI.
Table 3. Thermal properties of prepared resins.
| Tonset, °C | Hp*, J/g | Tp*, °C | |
|---|---|---|---|
| VBBr | 336 | 10.1 | 80.0 |
| VBBr-D3EI | 241 | 30.5 | 90.9 |
| VBBr-D4EI | 244 | 36.4 | 94.9 |
| VBC | 382 | 23.1 | 83.9 |
| VBC-D3EI | 174 | 4.7 | 66.0 |
| VBC-D4EI | 213 | 6.7 | 69.3 |
| Lewatit TP 207 | 80 | ||
| Amberlyst 15 | 150 |
Tonset—temperature of sorbents decomposition determined from TGA measurements. Hp*—the heat of solvent evaporation. Tp*—the temperature at the maximum of the peak of Hp determined from DSC thermograms.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27051723
This entry is offline, you can click here to edit this entry!