High mobility group box 1 (HMGB1) is a highly conserved DNA-shepherding protein that is plentiful in the cell nucleus. HMGB1 is actively secreted by multiple cell types, including macrophages, monocytes, dendritic cells, natural killer cells, endothelial cells, and platelets, and passively by necrotic and damaged cells. Either mode can release substantial amounts of extracellular HMGB1, which participates in multiple biological functions. Serum and liver levels of HMGB1 are significantly increased in some schistosomiasis patients with inflammatory responses, suggesting a close association with disease progression.
- high mobility group protein box 1
- inflammation
1. Introduction
High mobility group box 1 (HMGB1) is a highly conserved DNA-shepherding protein that is plentiful in the cell nucleus [1]. HMGB1 is actively secreted by multiple cell types, including macrophages, monocytes, dendritic cells, natural killer cells, endothelial cells, and platelets [2], and passively by necrotic and damaged cells [3][4]. Either mode can release substantial amounts of extracellular HMGB1, which participates in multiple biological functions. Serum and liver levels of HMGB1 are significantly increased in some schistosomiasis patients with inflammatory responses, suggesting a close association with disease progression [5]. Moreover, recent studies suggest that HMGB1 is significantly upregulated in patients with schistosome-induced hepatic fibrosis as well as animal models [6]. Meanwhile, inhibition of the translation and release of HMGB1 or blocking related signaling pathways via the HMGB1 receptor can protect against hepatic inflammation and fibrosis, suggesting that HMGB1 may be a vital factor in schistosome-induced liver disease as well as a potential therapeutic target [6][7][8].
2. HMGB1 Structure
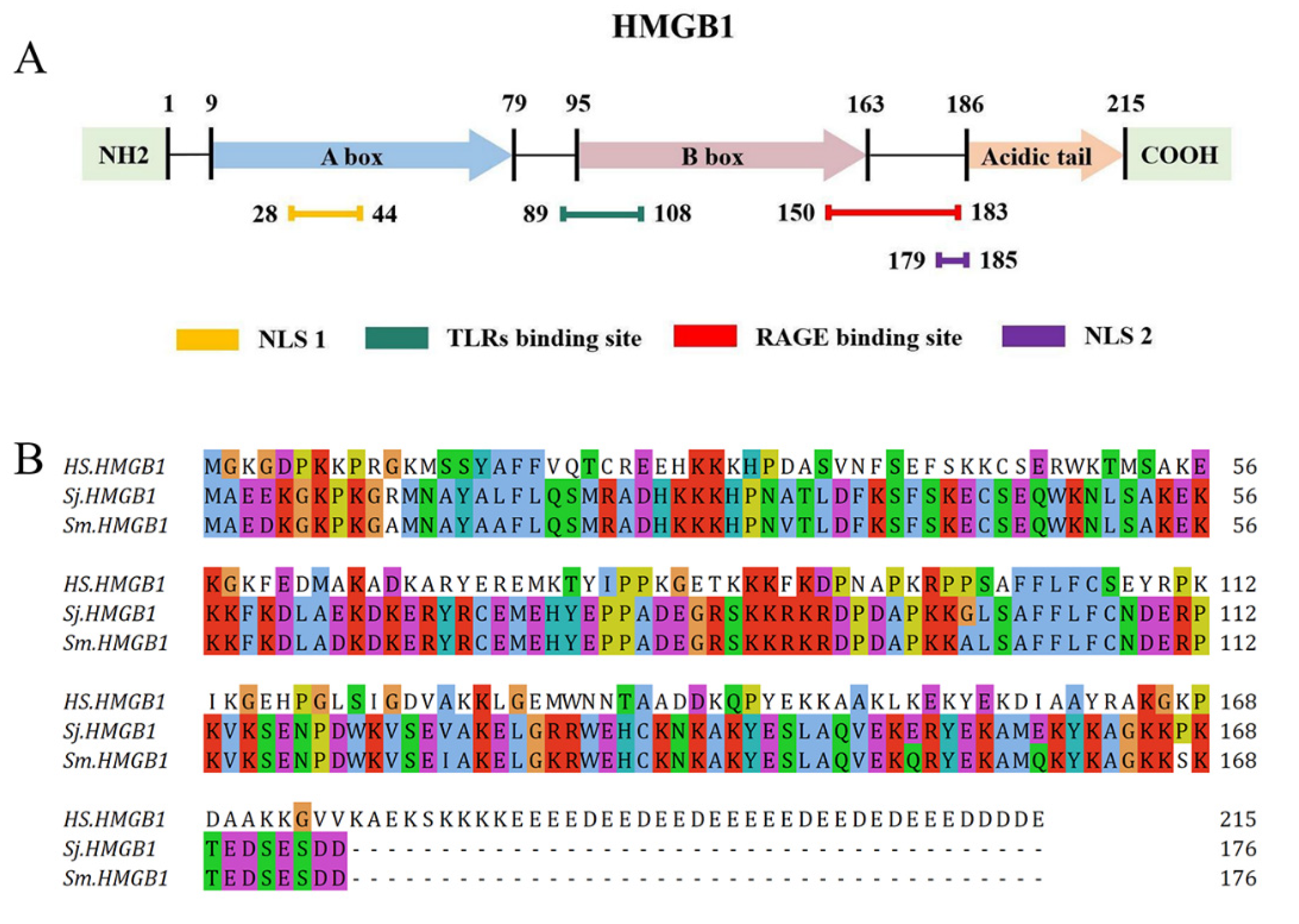
3. HMGB1 Receptors
4. HMGB1 as a Therapeutic Target
This entry is adapted from the peer-reviewed paper 10.3390/pathogens11030289
References
- Harris, H.E.; Andersson, U.; Pisetsky, D.S. HMGB1: A multifunctional alarmin driving autoimmune and inflammatory disease. Nat. Rev. Rheumatol. 2012, 8, 195–202.
- Hu, Y.; Wang, X.; Wei, Y.; Liu, H.; Zhang, J.; Shen, Y.; Cao, J. Functional Inhibition of Natural Killer Cells in a BALB/c Mouse Model of Liver Fibrosis Induced by Schistosoma japonicum Infection. Front. Cell. Infect. Microbiol. 2020, 10, 598987.
- Andersson, U.; Wang, H.; Palmblad, K.; Aveberger, A.C.; Bloom, O.; Erlandsson-Harris, H.; Janson, A.; Kokkola, R.; Zhang, M.; Yang, H.; et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J. Exp. Med. 2000, 192, 565–570.
- Andersson, U.; Tracey, K.J. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu. Rev. Immunol. 2011, 29, 139–162.
- Vicentino, A.R.R.; Carneiro, V.C.; Allonso, D.; Guilherme, R.F.; Benjamim, C.F.; Dos Santos, H.A.M.; Xavier, F.; Pyrrho, A.D.S.; Gomes, J.A.S.; Fonseca, M.C.; et al. Emerging Role of HMGB1 in the Pathogenesis of Schistosomiasis Liver Fibrosis. Front. Immunol. 2018, 9, 1979.
- Chen, H.; Li, G.; Zhang, J.; Zheng, T.; Chen, Q.; Zhang, Y.; Yang, F.; Wang, C.; Nie, H.; Zheng, B.; et al. Sodium butyrate ameliorates Schistosoma japonicum-induced liver fibrosis by inhibiting HMGB1 expression. Exp. Parasitol. 2021, 231, 108171.
- Ge, W.-S. Inhibition of high-mobility group box 1 expression by siRNA in rat hepatic stellate cells. World J. Gastroenterol. 2011, 17, 4090.
- Huang, H.; Nace, G.W.; McDonald, K.A.; Tai, S.; Klune, J.R.; Rosborough, B.R.; Ding, Q.; Loughran, P.; Zhu, X.; Beer-Stolz, D.; et al. Hepatocyte-specific high-mobility group box 1 deletion worsens the injury in liver ischemia/reperfusion: A role for intracellular high-mobility group box 1 in cellular protection. Hepatology 2014, 59, 1984–1997.
- Goodwin, G.H.; Sanders, C.; Johns, E.W. A new group of chromatin-associated proteins with a high content of acidic and basic amino acids. Eur. J. Biochem. 1973, 38, 14–19.
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342.
- Yang, H.; Antoine, D.J.; Andersson, U.; Tracey, K.J. The many faces of HMGB1: Molecular structure-functional activity in inflammation, apoptosis, and chemotaxis. J. Leukoc. Biol. 2013, 93, 865–873.
- de Oliveira, F.M.; de Abreu da Silva, I.C.; Rumjanek, F.D.; Dias-Neto, E.; Guimarães, P.E.; Verjovski-Almeida, S.; Stros, M.; Fantappié, M.R. Cloning the genes and DNA binding properties of High Mobility Group B1 (HMGB1) proteins from the human blood flukes Schistosoma mansoni and Schistosoma japonicum. Gene 2006, 377, 33–45.
- Neeper, M.; Schmidt, A.M.; Brett, J.; Yan, S.D.; Wang, F.; Pan, Y.C.; Elliston, K.; Stern, D.; Shaw, A. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J. Biol. Chem. 1992, 267, 14998–15004.
- Fritz, G. RAGE: A single receptor fits multiple ligands. Trends Biochem. Sci. 2011, 36, 625–632.
- Bierhaus, A.; Nawroth, P.P. Multiple levels of regulation determine the role of the receptor for AGE (RAGE) as common soil in inflammation, immune responses and diabetes mellitus and its complications. Diabetologia 2009, 52, 2251–2263.
- Taguchi, A.; Blood, D.C.; del Toro, G.; Canet, A.; Lee, D.C.; Qu, W.; Tanji, N.; Lu, Y.; Lalla, E.; Fu, C.; et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000, 405, 354–360.
- Huttunen, H.J.; Kuja-Panula, J.; Rauvala, H. Receptor for advanced glycation end products (RAGE) signaling induces CREB-dependent chromogranin expression during neuronal differentiation. J. Biol. Chem. 2002, 277, 38635–38646.
- Huttunen, H.J.; Fages, C.; Rauvala, H. Receptor for advanced glycation end products (RAGE)-mediated neurite outgrowth and activation of NF-kappaB require the cytoplasmic domain of the receptor but different downstream signaling pathways. J. Biol. Chem. 1999, 274, 19919–19924.
- Yan, S.D.; Schmidt, A.M.; Anderson, G.M.; Zhang, J.; Brett, J.; Zou, Y.S.; Pinsky, D.; Stern, D. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J. Biol. Chem. 1994, 269, 9889–9897.
- Huang, J.S.; Guh, J.Y.; Chen, H.C.; Hung, W.C.; Lai, Y.H.; Chuang, L.Y. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J. Cell. Biochem. 2001, 81, 102–113.
- Yamagishi, S.; Matsui, T. Role of receptor for advanced glycation end products (RAGE) in liver disease. Eur. J. Med. Res. 2015, 20, 15.
- Gaens, K.H.; Niessen, P.M.; Rensen, S.S.; Buurman, W.A.; Greve, J.W.; Driessen, A.; Wolfs, M.G.; Hofker, M.H.; Bloemen, J.G.; Dejong, C.H.; et al. Endogenous formation of Nε-(carboxymethyl)lysine is increased in fatty livers and induces inflammatory markers in an in vitro model of hepatic steatosis. J. Hepatol. 2012, 56, 647–655.
- Nomoto, K.; Tsuneyama, K.; Abdel Aziz, H.O.; Takahashi, H.; Murai, Y.; Cui, Z.G.; Fujimoto, M.; Kato, I.; Hiraga, K.; Hsu, D.K.; et al. Disrupted galectin-3 causes non-alcoholic fatty liver disease in male mice. J. Pathol. 2006, 210, 469–477.
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511.
- Meng, X.; Zhang, X.; Su, X.; Liu, X.; Ren, K.; Ning, C.; Zhang, Q.; Zhang, S. Daphnes Cortex and its licorice-processed products suppress inflammation via the TLR4/NF-κB/NLRP3 signaling pathway and regulation of the metabolic profile in the treatment of rheumatoid arthritis. J. Ethnopharmacol. 2022, 283, 114657.
- Cai, J.; Li, J.; Zhou, Y.; Wang, J.; Li, J.; Cui, L.; Meng, X.; Zhu, G.; Wang, H. Staphylococcus aureus facilitates its survival in bovine macrophages by blocking autophagic flux. J. Cell. Mol. Med. 2020, 24, 3460–3468.
- Liu, K.; Ding, T.; Fang, L.; Cui, L.; Li, J.; Meng, X.; Zhu, G.; Qian, C.; Wang, H.; Li, J. Organic Selenium Ameliorates Staphylococcus aureus-Induced Mastitis in Rats by Inhibiting the Activation of NF-κB and MAPK Signaling Pathways. Front. Vet. Sci. 2020, 7, 443.
- Li, Y.; Xu, B.; Yang, J.; Wang, L.; Tan, X.; Hu, X.; Sun, L.; Chen, S.; Zhu, L.; Chen, X.; et al. Liraglutide protects against lethal renal ischemia-reperfusion injury by inhibiting high-mobility group box 1 nuclear-cytoplasmic translocation and release. Pharmacol. Res. 2021, 173, 105867.
- Azam, S.; Jakaria, M.; Kim, I.S.; Kim, J.; Haque, M.E.; Choi, D.K. Regulation of Toll-Like Receptor (TLR) Signaling Pathway by Polyphenols in the Treatment of Age-Linked Neurodegenerative Diseases: Focus on TLR4 Signaling. Front. Immunol. 2019, 10, 1000.
- Guo, Q.; Qu, H.; Zhang, H.; Zhong, X. Prunella vulgaris L. Attenuates Experimental Autoimmune Thyroiditis by Inhibiting HMGB1/TLR9 Signaling. Drug Des. Dev. Ther. 2021, 15, 4559–4574.
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Balasubramaniam, V.; Othman, I.; Shaikh, M.F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur. J. Pharmacol. 2019, 858, 172487.
- Yu, R.; Jiang, S.; Tao, Y.; Li, P.; Yin, J.; Zhou, Q. Inhibition of HMGB1 improves necrotizing enterocolitis by inhibiting NLRP3 via TLR4 and NF-kappaB signaling pathways. J. Cell. Physiol. 2019, 234, 13431–13438.
- Khambu, B.; Yan, S.; Huda, N.; Yin, X.M. Role of High-Mobility Group Box-1 in Liver Pathogenesis. Int. J. Mol. Sci. 2019, 20, 5314.
- Wyganowska-Swiatkowska, M.; Nohawica, M.; Grocholewicz, K.; Nowak, G. Influence of Herbal Medicines on HMGB1 Release, SARS-CoV-2 Viral Attachment, Acute Respiratory Failure, and Sepsis. A Literature Review. Int. J. Mol. Sci. 2020, 21, 4639.
- Wei, J.; Alfajaro, M.M.; DeWeirdt, P.C.; Hanna, R.E.; Lu-Culligan, W.J.; Cai, W.L.; Strine, M.S.; Zhang, S.M.; Graziano, V.R.; Schmitz, C.O.; et al. Genome-wide CRISPR Screens Reveal Host Factors Critical for SARS-CoV-2 Infection. Cell 2021, 184, 76–91.e13.
- Chen, X.; Ling, Y.; Wei, Y.; Tang, J.; Ren, Y.; Zhang, B.; Jiang, F.; Li, H.; Wang, R.; Wen, W.; et al. Dual regulation of HMGB1 by combined JNK1/2-ATF2 axis with miR-200 family in nonalcoholic steatohepatitis in mice. FASEB J. 2018, 32, 2722–2734.
- Entezari, M.; Weiss, D.J.; Sitapara, R.; Whittaker, L.; Wargo, M.J.; Li, J.; Wang, H.; Yang, H.; Sharma, L.; Phan, B.D.; et al. Inhibition of high-mobility group box 1 protein (HMGB1) enhances bacterial clearance and protects against Pseudomonas Aeruginosa pneumonia in cystic fibrosis. Mol. Med. 2012, 18, 477–485.