Cutaneous Melanoma (CM) is an aggressive and invasive cancer of the skin. Epigenetic mechanisms are fundamentally important for cancer initiation and development. The application of some insights may contribute to further progress in the diagnosis and therapy of melanoma, a deadly type of cancer.
- melanoma
- DNA methylation
- epigenetics
1. Etiology of Cutaneous Melanoma
2. Histone Modifications and Significance for Melanoma Progression
3. DNA Methylation Alterations in Melanoma
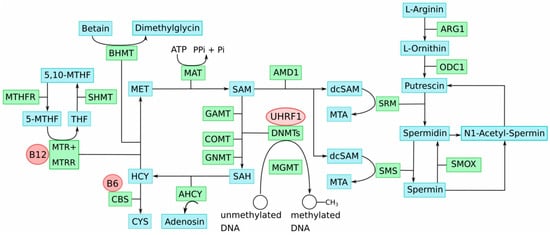
4. One-Carbon Metabolism in the Etiology of Epigenomic Aberrations in Melanoma
While a plethora of DNA methylation alterations occur in melanoma, it remains elusive how they are caused. It has been proposed that either active processes, e.g., an aberrant activity or function of DNMT enzymes, or passive ones, for instance, changes in epigenetic modifications that regulate targeting of DNA methylation, may be involved [30].
A major risk factor for bladder cancer is persistent exposure to the harmful carcinogens of tobacco smoking, which is estimated to account for 50% of tumors [31] and contributes to the high mutational rate in that cancer. There is thus a parallel to the etiology of melanoma, where exposure to another exogenous carcinogen—harmful UVB radiation—is the major cause. The previously proposed PrimeEpiHit hypothesis for UC [31][32] may, therefore, explain methylation alterations in CM carcinogenesis. According to this modified hypothesis, chronic UVB radiation exposure may occasionally also hit genes with key functions in one-carbon-group metabolism. As a result, their transcription may become impaired, and subsequently, their epigenetic status may be altered. Epigenetic silencing because of gene disruption has been experimentally demonstrated [33]. Interestingly, analyzing a comprehensive mortality rate dataset for 30 types of cancer for 52 provinces in Spain, spanning 1978–1992, it has been found that melanoma correlated with bladder and lung cancer, suggesting common risk factors and mechanisms [34]. Key genes involved in one-carbon-group metabolism of course comprise a very small percentage of the whole genome, but due to the chronic carcinogen exposure, nonetheless, this may occur at some minor frequency, which appears in accordance with the low incidence.
5. Epigenetic Diagnosis Based on Differential Chromatin Organization and DNA Methylation in Melanoma
Focusing first on cancer-specific differential chromatin organization, for taking advantage of it in cancer diagnosis and therapy, it suggests screening after sites of chromatin with distinct differential organized structures for various melanoma stages, e.g., the first mesenchymal stage, which arises after the epithelial–mesenchymal transition. Once detecting such promising chromatin areas with, e.g., a more relaxed chromatin organization in a pathogenic melanoma cell population, it would be able to target it either for diagnosis or even for therapy. For diagnosis, e.g., taking advantage of the limited effectiveness of micrococcus nuclease (MN) on closed chromatin, it would be possible to pretreat melanoma and reference DNA and provide cancer-cell-originated DNA templates of MN-affected integrity for assaying them by sensitive PCR assays, as previously demonstrated [39]. For therapy, those cancer-cell-specific, differential organized chromatin areas could be targeted by CRISPR/Cas in vivo, taking advantage of its selective preference feature. Evolved as an adaptive immunity system that protects bacteria and archaea against phages and plasmids [40] by directing sequence-specific Cas9 endonuclease-mediated double-strand DNA cleavage (DSB) to the intruder’s DNA, and hence destroying its genetic information [41], this system has evolutionarily never been encountered as a substrate DNA, which is organized in complex, high-order, structured chromatin and, hence, has a high preference for binding to more easily accessible chromatin regions [42][43][44].
A further basic approach is to find within a certain distinct cancer cell population of interest, differentially methylated CpG dinucleotides forming a distinct, consistent, and characteristic methylation signature for this cell population. In the former investigations on prostate cancer and urothelial cancer specimens from patients, it was able to discover such unique DNA methylation signatures. The proposed strategy [32] is to screen using DNA methylation array technology after such differentially methylated CpG regions, and once detected, to validate and precisely dissect the cancer cell-specific DNA methylation pattern by bisulfite genomic sequencing [45]. Afterward, based on this information, it can define the outmost most suitable and robust MSPCR primers to sensitively detect this methylation signature.
Furthermore, a new methodological approach to separate cell-free DNA from cellular DNA and unreservedly apply bisulfite genomic sequencing and MSPCR on this pure cell-free DNA is recently presented. It is established that all tumors shed their cell-free DNAs that bear their unique DNA methylation patterns into the bloodstream [45], hence providing an easily accessible and exciting Achilles’ heel of cancer for effective diagnosis. For instance, PTEN promoter methylation was reported in cell-free DNA from 62% of the melanoma serum samples examined by pyrosequencing, indicating a good correlation with the same epigenetic alteration found in paired melanoma tissues [46].
MSPCR has been improved and enables, for the first time, relative quantification of DNA methylation in samples with identical and unequal genetic settings [47]. This is of importance for cancer samples, which often yield genetic aberrations, including melanoma, characterized by a higher number of chromosomal structural aberrations [48], and bladder carcinoma, characterized by early (pTa, pT1) chromosomal changes and imbalances [49]. A conventional normalization of MSPCR by using a housekeeping gene such as GAPDH or the ACTB gene may result in false results, especially if tumor samples from different patients must be compared, because, e.g., the copy number of the DNA segment containing the housekeeping gene chosen for normalization may vary in cancer DNA. The reliable measurement of DNA methylation by using MSPCR is based on DNA amplification, and it is extremely hindered by a variable number of template segments. In contrast, by using the idiolocal normalization of real-time, methylation-specific PCR (IDLN–MSP) [47], the normalization loci are chosen adjacent to the targeted loci. With this approach, it is guaranteed that in real-time MSPCR, the number of normalization loci is always as high as the number of targeted loci, reducing false-negative results in a significant manner, and this contributes to a significant improvement of DNA methylation measurements by MSPCR in tumor samples of different patient origin [47].
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031531
References
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Stratton, M.R.; et al. Signatures of mutational processes in human cancer. Nature 2013, 22, 415–421.
- Dimitriou, F.; Krattinger, R.; Ramelyte, E.; Barysch, M.J.; Micaletto, S.; Dummer, R.; Goldinger, S.M. The World of Melanoma: Epidemiologic, Genetic, and Anatomic Differences of Melanoma Across the Globe. Curr. Oncol. Rep. 2018, 24, 87.
- DeLeon, T.T.; Almquist, D.R.; Kipp, B.R.; Langlais, B.T.; Mangold, A.; Winters, J.L.; Kosiorek, H.E.; Joseph, R.W.; Dronca, S.R.; Bryce, A.H.; et al. Assessment of clinical outcomes with immune checkpoint inhibitor therapy in melanoma patients with CDKN2A and TP53 pathogenic mutations. PLoS ONE 2020, 20, e0230306.
- Colombino, M.; Capone, M.; Lissia, A.; Cossu, A.; Rubino, C.; De Giorgi, V.; Massi, D.; Fonsatti, E.; Staiban, S.; Palmieri, G.; et al. BRAF/NRAS mutation frequencies among primary tumors and metastases in patients with melanoma. J. Clin. Oncol. 2012, 10, 2522–2529.
- Ichihashi, M.; Ueda, M.; Budiyanto, A.; Bito, T.; Oka, M.; Fukunaga, M.; Tsuru, K.; Horikawa, T. UV-induced skin damage. Toxicology 2003, 189, 21–39.
- Uzdensky, A.; Demyanenko, S.; Bibov, M.; Sharifulina, S.; Kit, O.; Przhedetski, Y.; Pozdnyakova, V. Expression of proteins involved in epigenetic regulation in human cutaneous melanoma and peritumoral skin. Tumor Biol. 2014, 35, 8225–8233.
- Smallwood, A.; Estève, P.O.; Pradhan, S.; Carey, M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007, 21, 1169–1178.
- Orouji, E.; Utikal, J. Tackling malignant melanoma epigenetically: Histone lysine methylation. Clin. Epigenetics 2018, 10, 145.
- Dang, N.N.; Jiao, J.; Meng, X.; An, Y.; Han, C.; Huang, S. Abnormal overexpression of G9a in melanoma cells promotes cancer progression via upregulation of the Notch1 signaling pathway. Aging 2020, 12, 2393–2407.
- Miura, S.; Maesawa, C.; Shibazaki, M.; Yasuhira, S.; Kasai, S.; Tsunoda, K.; Maeda, F.; Takahashi, K.; Akasaka, T.; Masuda, T. Immunohistochemistry for histone h3 lysine 9 methyltransferase and demethylase proteins in human melanomas. Am. J. Dermatopathol. 2014, 36, 211–216.
- Tan, Y.; Tajik, A.; Chen, J.; Jia, Q.; Chowdhury, F.; Wang, L.; Chen, J.; Zhang, S.; Hong, Y.; Yi, H.; et al. Matrix softness regulates plasticity of tumour-repopulating cells via H3K9 demethylation and Sox2 expression. Nat. Commun. 2014, 5, 4619.
- Kelly, G.M.; Al-Ejeh, F.; McCuaig, R.; Casciello, F.; Ahmad Kamal, N.; Ferguson, B.; Pritchard, A.L.; Ali, S.; Silva, I.P.; Wilmott, J.S.; et al. G9a Inhibition Enhances Checkpoint Inhibitor Blockade Response in Melanoma. Clin. Cancer Res. 2021, 27, 2624–2635.
- Ceol, C.J.; Houvras, Y.; Jane-Valbuena, J.; Bilodeau, S.; Orlando, D.A.; Battisti, V.; Fritsch, L.; Lin, W.M.; Hollmann, T.J.; Ferré, F.; et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature 2011, 471, 513–517.
- Kim, G.; Kim, J.Y.; Lim, S.C.; Lee, K.Y.; Kim, O.; Choi, H.S. SUV39H1/DNMT3A-dependent methylation of the RB1 promoter stimulates PIN1 expression and melanoma development. FASEB J. 2018, 32, 5647–5660.
- Vélez-Cruz, R.; Johnson, D.G. The Retinoblastoma (RB) Tumor Suppressor: Pushing Back against Genome Instability on Multiple Fronts. Int. J. Mol. Sci. 2017, 18, 1776.
- Pützer, B.M.; Steder, M.; Alla, V. Predicting and preventing melanoma invasiveness: Advances in clarifying E2F1 function. Expert Rev. Anticancer Ther. 2010, 10, 1707–1720.
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151.
- Sarkar, D.; Leung, E.Y.; Baguley, B.C.; Finlay, G.J.; Askarian-Amiri, M.E. Epigeneticregulation in human melanoma: Past and future. Epigenetics 2015, 10, 103–121.
- de Sá, B.C.S.; Fugimori, M.L.; Ribeiro, K.d.C.B.; Duprat Neto, J.P.; Neves, R.I.; Landman, G. Proteins involved in pRb and p53 pathways are differentially expressed in thin and thick superficial spreading melanomas. Melanoma Res. 2009, 19, 135–141.
- Straume, O.; Smeds, J.; Kumar, R.; Hemminki, K.; Akslen, L.A. Significant impact of promoter hypermethylation and the 540 C>T polymorphism of CDKN2A in cutaneous melanoma of the vertical growth phase. Am. J. Pathol. 2002, 161, 229–237.
- Tiffen, J.; Gallagher, S.J.; Filipp, F.; Gunatilake, D.; Emran, A.A.; Cullinane, C.; Dutton-Register, K.; Aoude, L.; Hayward, N.; Chatterjee, A.; et al. EZH2 Cooperates with DNA Methylation to Downregulate Key Tumor Suppressors and IFN Gene Signatures in Melanoma. J. Investig. Dermatol. 2020, 140, 2442–2454.e5.
- Moran, B.; Silva, R.; Perry, A.S.; Gallagher, W.M. Epigenetics of malignant melanoma. Semin. Cancer Biol. 2018, 51, 80–88.
- Bachmann, I.M.; Halvorsen, O.J.; Collett, K.; Stefansson, I.M.; Straume, O.; Haukaas, S.A.; Salvesen, H.B.; Otte, A.P.; Akslen, L.A. EZH2 expression is associated with high proliferation rate and aggressive tumor subgroups in cutaneous melanoma and cancers of the endometrium, prostate, and breast. J. Clin. Oncol. 2006, 24, 268–273.
- Sengupta, D.; Byrum, S.D.; Avaritt, N.L.; Davis, L.; Shields, B.; Mahmoud, F.; Reynolds, M.; Orr, L.M.; Mackintosh, S.G.; Shalin, S.C.; et al. Quantitative Histone Mass Spectrometry Identifies Elevated Histone H3 Lysine27 (Lys27) Trimethylation in Melanoma. Mol. Cell Proteom. 2016, 15, 765–775.
- Tellez, C.S.; Shen, L.; Estécio, M.R.H.; Jelinek, J.; Gershenwald, J.E.; Issa, J.P.J. CpG island methylation profiling in human melanoma cell lines. Melanoma Res. 2009, 19, 146–155.
- Conway, K.; Edmiston, S.N.; Khondker, Z.S.; Groben, P.A.; Zhou, X.; Chu, H.; Kuan, P.F.; Hao, H.; Carson, C.; Berwick, M.; et al. DNA-methylation profiling distinguishes malignant melanomas from benign nevi. Pigment Cell Melanoma Res. 2011, 24, 352–360.
- Akbani, R.; Akdemir, K.C.; Aksoy, B.A.; Albert, M.; Ally, A.; Amin, S.B.; Arachchi, H.; Arora, A.; Auman, J.T.; Kwong, L.N.; et al. Genomic Classification of Cutaneous Melanoma. Cell 2015, 161, 1681–1696.
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7, 16.
- Ecsedi, S.I.; Hernandez-Vargas, H.; Lima, S.C.; Herceg, Z.; Adany, R.; Balazs, M. Transposable hypomethylation is associated with metastatic capacity of primary melanomas. Int. J. Clin. Exp. Path. 2013, 6, 2943–2948.
- Micevic, G.; Theodosakis, N.; Bosenberg, M. Aberrant DNA methylation in melanoma: Biomarker and therapeutic opportunities. Clin. Epigenet. 2017, 9, 34.
- Erichsen, L.; Ghanjati, F.; Beermann, A.; Poyet, C.; Hermanns, T.; Schulz, W.A.; Seifert, H.H.; Wild, P.J.; Buser, L.; Kröning, A.; et al. Aberrantmethylated key genes of methyl group metabolism within the molecularetiology of urothelial carcinogenesis. Sci. Rep. 2018, 8, 3477.
- Erichsen, L.; Seifert, H.H.; Schulz, W.A.; Hoffmann, M.J.; Niegisch, G.; Araúzo-Bravo, M.J.; Bendhack, M.L.; Poyet, C.; Hermanns, T.; Beermann, A.; et al. Basic Hallmarks of Urothelial Cancer Unleashed in Primary Uroepithelium by Interference with the Epigenetic Master Regulator ODC1. Sci. Rep. 2020, 10, 3808.
- Castanotto, D.; Tommasi, S.; Li, M.; Li, H.; Yanow, S.; Pfeifer, G.P.; Rossi, J.J. Short hairpin RNA-directed cytosine (CpG) methylation of the RASSF1A gene promoter in HeLa cells. Mol. Ther. 2005, 12, 179–183.
- Grant, W.B. An ecologic study of cancer mortality rates in Spain with respect to indices of solar UVB irradiance and smoking. Int. J. Cancer 2007, 120, 1123–1128.
- Hoffman, D.R.; Cornatzer, W.E.; Duerre, J.A. Relationship between tissue levels of S-adenosylmethionine, S-adenylhomocysteine, and transmethylation reactions. Can. J. Biochem. 1979, 57, 56–65.
- Ulrey, C.L.; Liu, L.; Andrews, L.G.; Tollefsbol, T.O. The impact of metabolism on DNA methylation. Hum. Mol. Genet. 2005, 14, R139–R147.
- Coppedè, F. One-carbon metabolism and Alzheimer’s disease: Focus on epigenetics. Curr. Genomics 2010, 11, 246–260.
- Pegg, A.E. Spermidine/spermine-N(1)-acetyltransferase: A key metabolic regulator. Am. J. Physiol. Endocrinol. Metab. 2008, 29, E995–E1010.
- Graffmann, N.; Santourlidis, S.; Christ, J.; Wernet, P.; Uhrberg, M. Direct and quantitative analysis of chromatin accessibility by MIRECAL–a Micrococcusnuclease/real-time PCR chromatin accessibility assay with locus specificity. Anal. Biochem. 2006, 354, 308–310.
- Deltcheva, E.; Chylinski, K.; Sharma, C.M.; Gonzales, K.; Chao, Y.; Pirzada, Z.A.; Eckert, M.R.; Vogel, J.; Charpentier, E. CRISPR RNA maturation bytrans-encoded small RNA and host factor RNase III. Nature 2011, 471, 602–607.
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821.
- Kuscu, C.; Arslan, S.; Singh, R.; Thorpe, J.; Adli, M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014, 32, 677–683.
- O’Geen, H.; Henry, I.M.; Bhakta, M.S.; Meckler, J.F.; Segal, D.J. A genome-wide analysis of Cas9 binding specificity using ChIP-seq and targeted sequence capture. Nucleic Acids Res. 2015, 43, 3389–3404.
- Wu, X.; Scott, D.A.; Kriz, A.J.; Chiu, A.C.; Hsu, P.D.; Dadon, D.B.; Cheng, A.W.; Trevino, A.E.; Konermann, S.; Chen, S.; et al. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014, 32, 670–676.
- Ghanjati, F.; Beermann, A.; Hermanns, T.; Poyet, C.; Araúzo-Bravo, M.J.; Seifert, H.H.; Schmidtpeter, M.; Goering, W.; Sorg, R.; Wernet, P.; et al. Unreserved application of epigenetic methods to define differences of DNAmethylation between urinary cellular and cell-free DNA. Cancer Biomark. 2014, 14, 295–302.
- Giunta, E.F.; Arrichiello, G.; Curvietto, M.; Pappalardo, A.; Bosso, D.; Rosanova, M.; Diana, A.; Giordano, P.; Petrillo, A.; Ottaviano, M.; et al. Epigenetic Regulation in Melanoma: Facts and Hopes. Cells 2021, 10, 2048.
- Santourlidis, S.; Ghanjati, F.; Beermann, A.; Hermanns, T.; Poyet, C. IDLN-MSP: Idiolocal normalization of real-time methylation-specific PCR for genetic imbalanced DNA specimens. Biotechniques 2016, 60, 84–87.
- Hayward, N.K.; Wilmott, J.S.; Waddell, N.; Johansson, P.A.; Field, M.A.; Nones, K.; Patch, A.M.; Kakavand, H.; Alexandrov, L.B.; Burke, H.; et al. Whole-genome landscapes of major melanoma subtypes. Nature 2017, 545, 175–180.
- Simon, R.; Bürger, H.; Brinkschmidt, C.; Böcker, W.; Hertle, L.; Terpe, H.J. Chromosomal aberrations associated with inasion in papillary superficial bladder cancer. J. Pathol. 1998, 185, 345–351.