The Ventx family is one of the subfamilies of the ANTP (antennapedia) superfamily and belongs to the NK-like (NKL) subclass. Ventx is a homeobox transcription factor and has a DNA-interacting domain that is evolutionarily conserved throughout vertebrates. It has been extensively studied in Xenopus, zebrafish, and humans. The Ventx family contains transcriptional repressors widely involved in embryonic development and tumorigenesis in vertebrates. Several studies have documented that the Ventx family inhibited dorsal mesodermal formation, neural induction, and head formation in Xenopus and zebrafish. Moreover, Ventx2.2 showed functional similarities to Nanog and Barx1, leading to pluripotency and neural-crest migration in vertebrates. Among them, Ventx protein is an orthologue of the Ventx family in humans. Studies have demonstrated that human Ventx was strongly associated with myeloid-cell differentiation and acute myeloid leukemia.
1. Introduction
Xenopus laevis is an established animal model for studying vertebrate development, neurobiology, immunology, cell biology, and toxicology [
1,
2].
Xenopus is a pseudo-tetraploidy organism [
3,
4]. Its genome tetraploidy arose from the merging of two progenitor ancestral frog genomes millions of years ago [
3,
4,
5,
6]. During genome evolution, the
Xenopus laevis genome was subdivided into a longer subgenome (L-subgenome) and a shorter subgenome (S-subgenome). There is some gene duplication. However, most of the functional genes are located in the L-subgenome, while the majority of non-functional genes or pseudogenes are in the S-subgenome, formed by either the deletion of exons or frameshifts by single-nucleotide insertion/deletion [
4,
5,
6,
7]. The presence of non-functional genes or pseudogenes leads to doses compensation. Many families of transcriptional factors have evolved in vertebrates to regulate a wide network of downstream genes in a temporospatial-dependent manner to orchestrate orderly embryonic development and organogenesis [
8,
9,
10]. Among those families, the homeobox domain-containing families, including the Ventx family, play crucial roles during embryonic development and are widely involved in tumorigenesis in vertebrates [
9,
11]. The Ventx family is a transcription-repressor family belonging to the NKL subclass. The NKL subclass is one of the subclasses of ANTP (the
antennapedia gene, one of the Hox genes identified within the ANT-C homeotic complex of Drosophila) superfamily [
8,
9,
11]. The Ventx family has been extensively studied in
Xenopus, zebrafish, and humans. BMP4/Smad1 and FGF/Xbra signaling initiates the expression of the Ventx family members in
Xenopus [
12,
13,
14,
15]. Recent studies have shown that BMP4/Smad1 and FGF/Xbra controlled the Ventx family expression in both
Xenopus [
13] and humans [
16,
17,
18]. BMP4/Smad1 is involved in a signaling path leading to the positive regulation of the expression of Vega family genes (a homolog of Ventx family in
Xenopus) in zebrafish [
19,
20,
21]. Smad1 and
Xenopus brachyury (Xbra) physically interact within the 5′-upstream regions of
ventx1.1 and
ventx2.1, either in combination or separately, to augment
ventx1.1 and
ventx2.1 transcription in
Xenopus [
13,
14,
16,
22,
23,
24,
25,
26]. In addition, Ventx2.2 is an ortholog of Nanog in
Xenopus and exhibited a sequential and functional similarity to Nanog and Barx1 [
27,
28]. Although studies have revealed that Ventx synteny has been evolutionarily conserved in cats (Chr28), humans (Chr10, q26.3 loci),
Gallus (Chr6), zebrafish (Chr13 and Chr10), and
Xenopus (Chr7), it is absent in rodents, including rats and mice (
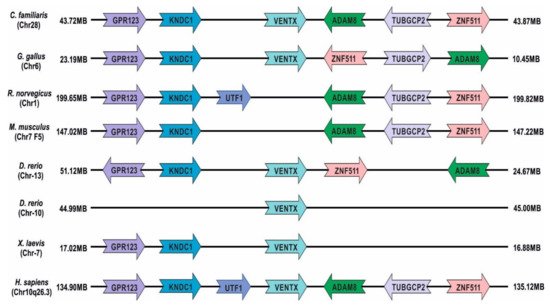
Figure 1) [
13,
29]. Ventx family genes are clustered at chromosome 7 in
Xenopus, though it has been identified as non-clustered in zebrafish (Chr13 and Chr10). In humans, seven pseudogenes of the Ventx family have been discovered, dispersed throughout different chromosomal locations, including VENTXP1 (Xp21.3), VENTXP2 (13q31.1), VENTXP3 (12q21.1), VENTXP4 (3p24.2), VENTXP5 (8p12), VENTXP6 (8q21.11), and VENTXP7 (3p24.3) [
11]. The Ventx family proteins play various roles during axial patterning, germ cell specification, and cell differentiation during the embryonic development of vertebrates [
30,
31,
32,
33,
34,
35,
36,
37,
38]. Additionally, the Ventx family members trigger ventral mesoderm, ectoderm, and ventral-blood-island (VBI) formations [
22,
30,
35,
39,
40,
41,
42]. In contrast, increased levels of the Ventx family directly suppresses the dorso-anterior development, inhibiting head formation and primary neurogenesis in
Xenopus and zebrafish [
15,
30,
33,
34,
38,
43,
44,
45]. In
Xenopus, the ectopic expression of Ventx family members attenuated the expression of organizer-specific genes, including
goosecoid (
gsc),
chordin,
noggin, and
foxd4l1.1b (an early neural marker); and the same occurred for
zic3 (a neuroectodermal marker) in
Xenopus embryos at St.11 [
13,
30,
33,
34,
35,
45,
46,
47]. This attenuation caused the inhibition of dorsal mesoderm formation and neuroectodermal induction. The suppressed expression of organizer-specific genes resulted in a headless phenotype. Studies have shown that Ventx2.2/Xom acts as a Nanog-like pluripotency marker in
Xenopus [
48,
49]. Overexpressed Ventx2.2 stabilizes the cells’ pluripotency and inhibits multiple-cell-lineage commitment during the embryonic development of
X. laevis.
Figure 1. Chromosomal localization of Ventx gene synteny in humans and other animal species relative to adjacent genes (color-coded).
Similarly to
Xenopus, BMP4/Smad1 controls the expression of the Vega family members [
34,
38,
45]. The ectopic expression of Vega family members diminishes the dorsal and organizer-specific markers, leading to embryonic ventralization. Studies have found elevated levels of Ventx in acute-myeloid-leukemia (AML) cells, macrophages, and dendritic cells to promote their differentiation and leukemogenesis in humans and animal models [
18,
32,
50,
51]. Studies have shown that p53 transactivates the expression of p21 (p21
Cip1/Waf1, an endogenous cell-cycle inhibitor) to inhibit the activity of cyclin–CDK complexes, including cyclin–Cdk1, cyclin–Cdk2, and cyclin–Cdk4/6 during G1 and S phases [
52,
53]. During DNA damage response, p53–p21 leads to cell senescence by inhibiting the CDK2/4–cyclinE/D1 complex, which arrests cell cycles. In contrast, Wu et al. (2011) revealed that Ventx directly transactivated the tumor suppression pathways in a p53–p21 and p16ink4a-Rb (a molecular link protein between cell senescence and tumor suppression)-dependent manner, leading to cellular senescence [
54]. These studies suggested that the Ventx family plays multifaceted roles in hematopoietic cell differentiation and AML progression, tumor suppression, and apoptotic-pathway activation. We review these roles of the Ventx family in
Xenopus, zebrafish, and humans and discuss the therapeutic potential of utilizing the Ventx family crosstalk pathways to inhibit cancer progression in humans. Additionally, we discuss the pseudo-tetraploidy, genome evolution, and gene duplication in
Xenopus,
Xenopus’s classes of the homeobox superfamily, and functional similarities of Ventx2.2 to Nanog and Barx1.
2. Human Ventx (VENT-like Homeobox Protein 2)
Ventx might be the only reported active member of the Ventx family in humans (
Figure 3A). It is also known as VENTX2 and HPX42B. Human Ventx showed 39%–67.66% similarity to Ventx family members in
Xenopus and zebrafish (
Table 3) [
17]. The ectopic expression of human Ventx in zebrafish embryos caused the loss of the anterior structure, resulting in the failure of notochord formation. In addition, overexpressed Ventx also leads to embryonic ventralization in
Xenopus and zebrafish. Similarly to the Ventx family in
Xenopus and zebrafish, the expression of human Ventx was also led by BMP4 signaling [
108]. Human Ventx works downstream of BMP4/Smad1 signaling and causes embryonic ventralization in
Xenopus and zebrafish. A study reported that Ventx was aberrantly expressed in CD34
+ acute myeloid leukemia (AML), and its maximum expression was found in CD33
+ myeloid cells but not in normal CD34
+/CD38
− leukemic cells [
18]. In addition, the abnormal expression of Ventx in normal CD34
+ cells could disturb the development of hematopoietic cells, leading to myeloid cell differentiation rather than lymphoid generation, both in vivo and in vitro. The shRNA-mediated knockdown of
Ventx resulted in the failure of AML cell proliferation [
18]. These studies indicated that Ventx may play a vital role in postnatal hematopoiesis and malignant myelopoiesis.
Ventx led to macrophage differentiation, depending on the cytokines involved, such as CSFs and IL-3. The inhibition of Ventx in primary monocytes suppressed monocyte-to-macrophage differentiation, suggesting that Ventx plays a significant role in macrophage differentiation and the ensuing activation of cytokines [
51]. A recent study has shown that Ventx was dramatically reduced in tumor-associated macrophages (TAMs), which are critical components of the tumor microenvironment (TME). The suppression of the immune system in TME was one of the key hurdles in developing effective immunotherapies to treat tumors [
109]. In addition, Ventx potentiated the cell growth and terminal differentiation of hematopoietic and immune cells in isolated samples of cancer patients. Moreover, Ventx regulated TAM plasticity, resulting in the M1-like phenotype of TAMs. Ventx suppressed the immune system in the TME, inhibited regulatory T-cell (Treg) differentiation, and promoted CD8 tumor-infiltrating lymphocytes [
109]. In addition, Ventx modulated TAM functions by inhibiting the immune suppression of TAM plasticity in the TME during tumorigenesis in colon-cancer models. The involvement of Ventx in macrophage differentiation and the suppression of TAM plasticity in TME indicates that Ventx could be a novel agent for targeting tumorigenesis. Recently, Le et al. (2020) showed that the expression level of Ventx was notably decreased in the pancreatic ductal adenocarcinoma-(PDA)-associated macrophages (TAMs) collected from verified human patients [
110]. The study showed that Ventx was necessary for regulating the phagocytosis, and the restored level of Ventx triggered phagocytosis by regulating the expression of Toll-like receptors 2/9, MyD88, and Mapk38 in PDA TAMs. Furthermore, the ectopic expression of Ventx inhibited the SHP1/2 phosphorylation level (the downstream molecules of SIRPa-CD47 signaling) to trigger phagocytosis. Ventx also induced phosphorylation focal adhesion kinase (FAK), leading to phagocytosis. Moreover, the elevated level of Ventx promoted the M1-like TAM phenotypes over M2-like TAM phenotypes to provide immunity against PDA by promoting the CD8 T-cell population over the Treg cell population in the PDA-associated tumor microenvironment. A study has shown that the M1/M2 and CD8/Treg ratio was associated with PDA prognosis [
111], and the increased population of M1-like TAMs promoted immunity against pancreatic cancers. These findings indicate that Ventx could be a valuable candidate to improve PDA prognosis through the promotion of M1-like and CD8 populations and phagocytosis in PDA.
An elevated level of Ventx was reported in immature human hematopoietic cells. The knockdown of the Ventx expression significantly expanded the population of human stem cells and multipotent progenitor cells (HSC/MPP) and promoted the survival of HSC, ex vivo [
112]. The suppression of
Ventx in grafted HSC/MPP cells also increased the engraftment potential of HSC/MPP in NOD/SCID/IL2Rγ2
null murine models. This suggested that Ventx could be a potent target for the application of HSC/MPP-based therapy. Recently, a study demonstrated that the expression of
Ventx robustly increased in normal myeloid cells and in AML patients [
32]. The overexpressed Ventx in myeloid cells reduced the expression of myeloid-terminal-differentiation-related genes in healthy CD34
+ stem and progenitor cells. Moreover, the transplantation of retrovirally-engineered Ventx-expressed bone marrow progenitor cells increased the population of primitive erythroid cells to trigger acute erythroid leukemia in transplanted mice. Furthermore, the concomitant expression of AML1-ETO (AML1-ETO, a fusion product of the chromosomal translocation t(8;21)(q22;q22)) usually occurs in acute myeloid leukemia (AML) with granulocytic differentiation, FAB M2 subtype [
113], and Ventx induced the expression of the erythroid markers, leading to AML in all transplanted mice. In addition, high levels of Ventx in human erythroleukemia patients significantly inhibited cell proliferation and tumor growth. However, Ventx acted as a tumor activator and triggered leukemogenesis, but some studies demonstrated its tumor-suppressant role in humans. A study reported its reduced expression in peripheral blood of chronic lymphocytic leukemia (CLL) cells and that it acted as a tumor suppressant [
99]. Ventx interacted with LEF/TCFs and inhibited the canonical Wnt/β-cat signaling (a positive regulator of cell proliferation) in CLL cells. Ventx and LEF/TCF interaction abrogates β-cat and LEF/TCFs complex formation and suppresss the proliferation-marker cyclin D1’s expression in CLL cells. Ventx-LEF/TCF interaction reduced the LEF/TCF-targeted oncogene, cyclin D1, to inhibit cyclin D1-mediated cell proliferation. The reduced level of Ventx in CLL cells promoted Wnt/β-cat and LEF/TCFs interaction, and in turn, induced cyclin D1 expression, leading to cell proliferation and cancer progression. Recently, it was reported that the differential methylation level of Ventx was significantly implicated in sarcoma progression in humans [
114]. A genome-wide methylation-profiling study in metastatic and recurrent sarcomas showed that Ventx was hypermethylated in metastasis pleomorphic rhabdomyosarcoma (Ple-RMS, a rare, aggressive sarcoma with poor prognosis), as compared to primary Ple-RMS. This study hypothesized that due to its involvement in tumor microenvironment and acting as an Wnt antagonist, Ventx may have strong therapeutic potential for sarcomas associated with immune cell content. A study showed that Ventx might also act as a negative regulator of tumor progression. Ventx activated both p16
ink4a-Rb and p53–p21 tumor-suppression pathways [
54]. Overexpressing Ventx led to a cell-senescence-like phenotype, followed by an irreversible cell-cycle arrest. Strikingly, the RNAi-mediated inhibition of
Ventx enhanced cell senescence and ameliorated the camptothecin (CPT) resistance in lymphocytic leukemia cells. Ventx-associated chemotherapeutic resistance and its involvement in the cell-senescence process suggested that Ventx could be a novel, potential target for cancer prevention. A recent study confirmed the anticancer role of Ventx and showed that Ventx triggered apoptosis in a p53-independent manner in cancer cells (HCT116 p53KO) but not in healthy cells, in vitro [
115]. In addition, Ventx activated the executioner caspase-3 in p53
−/− knockout cells, leading to the suppression of tumor growth.
Furthermore, the treatment of doxorubicin increased the level of Ventx in both p53 (WT) and p53−/− knockout cells. Ventx’s involvement in hematopoiesis, leukemogenesis, cell senescence, and macrophage differentiation is illustrated in Figure 4. These studies suggested a tumor-suppressant function of Ventx in both p53+/+ and p53−/− cells, and Ventx might be a novel therapeutic target for certain human cancers, working in a p53-independent manner. In summary, Ventx is deeply involved in hematopoiesis and leukemogenesis in humans, and it could be a future therapeutic target for combating tumorigenesis.
Nanog (Ventx-like) Is Considered an Oncogene in Humans
Nanog is a well-established stem-cell pluripotent marker and homeobox transcription factor which creates stem-cell-like properties in cancer cells [
92]. Earlier, we discussed the functional and homologic similarities between Ventx2.2 and Nanog and their roles in maintaining cell pluripotency and inhibiting differentiation. Cancer stem cells (CSCs) and normal stem cells can self-differentiate and self-renew. CSCs are immortal in nature, persistent in tumors, and provide nourishment for tumor maintenance, metastasize, and relapses [
116]. Similarly to Ventx, aberrant expression of Nanog was detected in a small subset of AML patients and an AML cell line, Namo-1 (serves as a model for AML) [
117]. Loss and gain of function studies have shown that Ventx, KLF4, Myb, and anti-apoptosis-factor MIR17HG are Nanog-targeted genes [
116]. These factors are widely involved in generating oncogenic properties and leading to leukemogenesis in humans. Nanog inhibited p53 in a Gli-MDM2-dependent manner in glioma tumors [
118,
119]. Nanog induced the expression of Gli1/2 (glioma-associated oncogenes) that is required for MDM2 activation, and inhibited p53 activity. The tumor suppressant p53 directly reduced the promoter activity of
Nanog (p53 is a direct inhibitor of Nanog). Nanog and p53 mutually inhibit each other expression in glioma tumors (for a review, see Grubelnik et al., 2020). However, p53 promoted Notch1 expression in early-stage oral squamous cell carcinoma. Notch1 is a positive regulator of Nanog [
120]. The Nanog-mediated inhibition of p53 indicated that Nanog was involved in cancer cell proliferation and tumor development in humans. Additionally, the p53-mediated inhibition of Nanog suggested that Nanog had oncogenic abilities and may promote the stem-cell-like nature of cancer cells, leading to cancer self-differentiation and self-renewal [
116,
120]. Meanwhile, the p53-mediated activation of Nanog in a Notch1-dependent manner indicated the importance of Nanog during embryonic development.
Although, Ventx synteny is absent in rodent, but a recent study has been demonstrated that Nanog acts like a Ventx ortholog in rodents [
121]. It showed that IGF2/IGF1R/Nanog signaling cascade regulates the proliferation of leukemia stem cells (LSCs) in mice. The overexpressed level of Nanog has been reported in CD34+ acute myeloid leukemia (AML) cells of patients and LSCs cells. Additionally, the knockdown of Nanog notably inhibits cell proliferation and induces cell cycle arrest and apoptosis. The si-mRNA-mediated silencing of Nanog suppresses the leukemogenesis of LSCs in mice. This report indicates that Nanog acts as a downstream target of IGF2/IGF1R signaling pathways that might positively regulate the Nanog transcription in LSCs. The overexpressed Nanog rescue colony formation ability of LSCs treated with IGF1R inhibitor (picropodophyllin). The knockdown of Nanog diminishes the colony formation ability of LSCs led by insulin-like growth factor 2 (IGF2) [
121]. These findings suggested that IGF2/IGF1R/Nanog signaling cascade plays a critical role in regulating the LSCs proliferation while the overexpressed levels of Nanog trigger AML proliferation in mice and leukemogenesis in mice LSCs. This study supports that Nanog may be a possible candidate for leukemogenesis in rodents and might act like a Ventx ortholog.
These studies showed that Nanog and Ventx not only share functional similarities in maintaining cell pluripotency to delay cell-lineage commitments during embryonic development, but they are also similarly involved in cancer progression and leukemogenesis in humans.
3. Current Insights
The Ventx and Vega families function downstream of BMP4/Smad1/5/8 signaling in vertebrates. BMP4/Smad1 upregulates the expression of
Ventx and
Vega families in the ventral regions of
Xenopus and zebrafish embryos, respectively. The Ventx family is known for its transcriptional repressor activity, which drives the proper axial formation during embryonic development. The Ventx family not only inhibited dorsally expressed genes but also activated the transcription of ventral specific genes, leading to axial patterning and cell-fate determination in zebrafish and
Xenopus [
12,
13,
14,
22,
34,
38,
45,
47,
71,
72,
83,
121]. Additionally, Ventx2.2 acted as a pluripotency regulator and maintained pluripotency in the VMZ. It could delay the blastula–gastrula transition during embryonic development of
Xenopus [
48,
49]. Additionally, the inhibition of Ventx2.2 may trigger multiple cell-lineage commitments and gastrulation in
Xenopus embryos.
In the Ventx family, the transactivation domain (TAD) of Ventx2.2 transactivated the Ventx genes, including
ventx1.1 and
ventx1.2, in a LEF1/TCF-dependent manner in
Xenopus [
14,
22,
24,
87]. The ectopic expression of Ventx2.2 inhibited β-cat levels, causing the induction of
gsk3β mRNA, leading to embryonic ventralization in
Xenopus [
105]. However, a study reported that Ventx2.2 was phosphorylated and degraded in a GSK3β-independent manner during gastrulation in
Xenopus [
86]. Ventx2.2 might be involved in GSK3b-mediated cranial neural-crest-cell (CNC) migration rather than β-cat-dependent CNC migration in
Xenopus. In humans, Ventx boosted cell proliferation and hematopoietic cell-terminal differentiation, including myeloid cell differentiation [
18,
32]. Studies have shown that Ventx also participated in monocyte–macrophage cell differentiation [
50,
51]. Ventx modulated TAM plasticity in TME to suppress the immune actions and macrophage differentiation against tumorigenesis [
109]. An elevated level of retrovirally-engineered Ventx caused erythroleukemia and expanded the abnormal primitive erythroid cells in bone marrow progenitor cells, leading to leukemic progression in transplanted mice [
32]. On the contrary, recent studies have revealed that Ventx may also act as an anti-cancer agent [
54,
115]. Ventx triggers apoptosis in p53
−/− knockout cell lines and activates cell senescence, which is followed by the activation of caspase-3, p53-p21, and p16
ink4-Rb pathways. In p53
−/− cells, doxorubicin augmented the Ventx level to trigger apoptosis in a caspase-3-dependent manner. Therefore, the Ventx family acts as a key player in dorsoventral axial patterning, pluripotency, cell-fate determination, hematopoiesis, apoptosis, and CNC migration. Additionally, the inhibition of Ventx family members suppresses cell proliferation and tumor growth in humans.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23052741