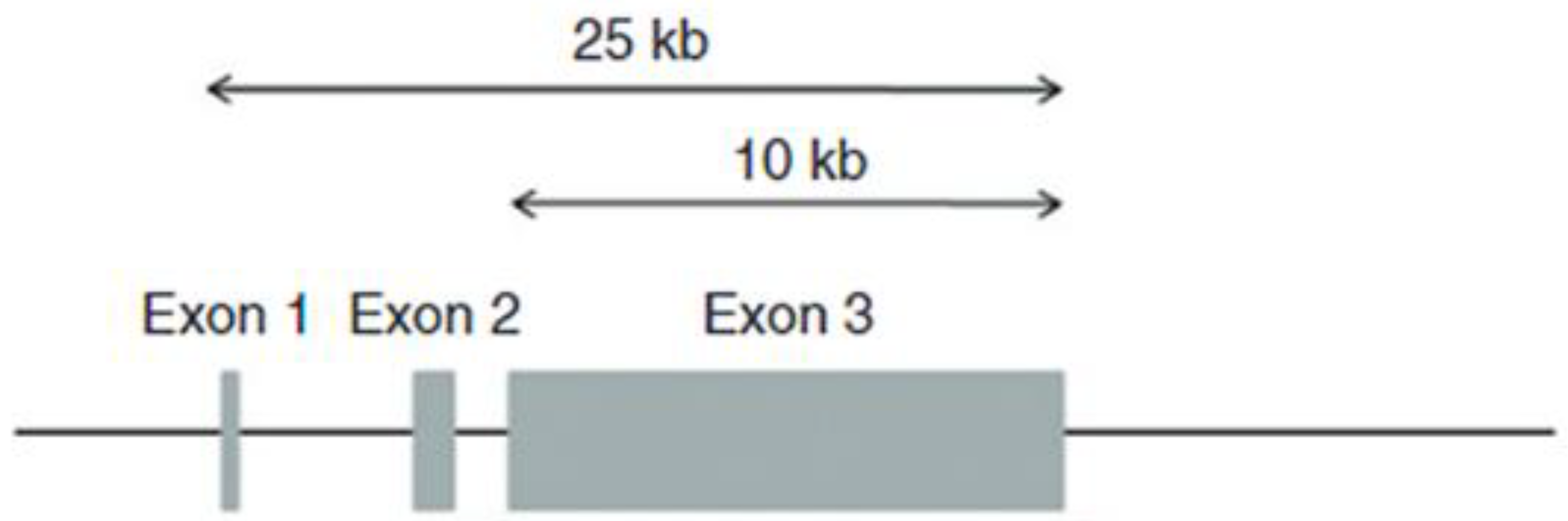
Keratohyalin granules were discovered in the mid-19th century in cells that terminally differentiate to form the outer, cornified layer of the epidermis. The first indications of the composition of these structures emerged in the 1960’s from a histochemical stain for histidine, followed by radioauto-graphic evidence for a high incidence of histidine incorporation into newly synthesized proteins in cells containing the granules. Research during the next three decades revealed the structure and function of the major protein in these granules, which was initially called the ‘histidine-rich pro-tein.’ Steinert and Dale named the protein ‘filaggrin’ in 1981 because of its ability to aggregate keratin intermediate filaments. The human gene for the precursor ‘profilaggrin’ was reported in 1991 to encode 10, 11 or 12 nearly identical repeats. Remarkably, the mouse and rat genes encode up to 20 repeats. The lifetime of filaggrin is the time required for keratinocytes in the granular layer to move into the inner cornified layer. During this transition, filaggrin facilitates the collapse of corneocytes into an impermeable surface barrier. The subsequent degradation of filaggrin is as remarkable as its synthesis and the end-products aide in maintaining moisture in the cornified layer. It became apparent that ichthyosis vulgaris and atopic dermatitis were associated with the absence of this protein. McLean’s team in 2006 identified the cause of these diseases by discov-ering loss-of-function mutations in the profilaggrin gene that led to dysfunction of the epidermal surface barrier. This story illustrates the complexity in maintaining a healthy, functional surface barrier.
- keratohyalin granules
- histidine-rich protein
- filaggrin
- profilaggrin
- ichthyosis vulgaris
- atopic dermatitis
- corneodesmosomes
- transglutaminase
1. Keratohyalin Granules and Histidine
Karen Holbrook [1] commented that “morphology is often the starting point of an investigation.” Developments in microscopy during the 19th century opened morphology of the cellular world to full view. The ability to thin-section fixed tissues and innovations in selectively staining cellular components provided biologists with opportunities to study structures of tissues and the organelles within cells. Several striking observations made by histochemical and radioautographic analyses of the epidermis were found, which led to the discovery of filaggrin and its short-lived but essential functions in assembly of a healthy surface barrier.
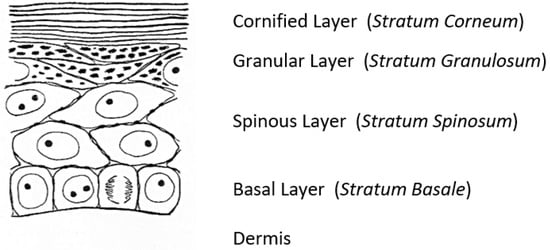
A fascinating morphological feature forms under the cornified layer of the epidermis (Figure 1) as keratinocytes move outward from the proliferative basal layer and terminally differentiate into corneocytes that form the surface barrier of the skin. Stephen Rothman [2], recounting the early history of studies on these ‘keratohyalin granules,’ remarked that during those slower years of “wax candles and horse carriages,” there was time for “detailed and precise observations that revealed the existence of granules in the granular layer, [which were] first observed and recorded by Auffhammer (1869).” The granules were further studied by Langerhans (1873) and designated ‘keratohyalin’ in 1882 by Waldeyer. Although very active in research on the skin throughout his career, Rothman commented shortly before he died in 1963 that not much more had been learned since those early days [3]. He did not live to see the dramatic developments emerging from the investigations of these unusual structures that were already underway in several laboratories.
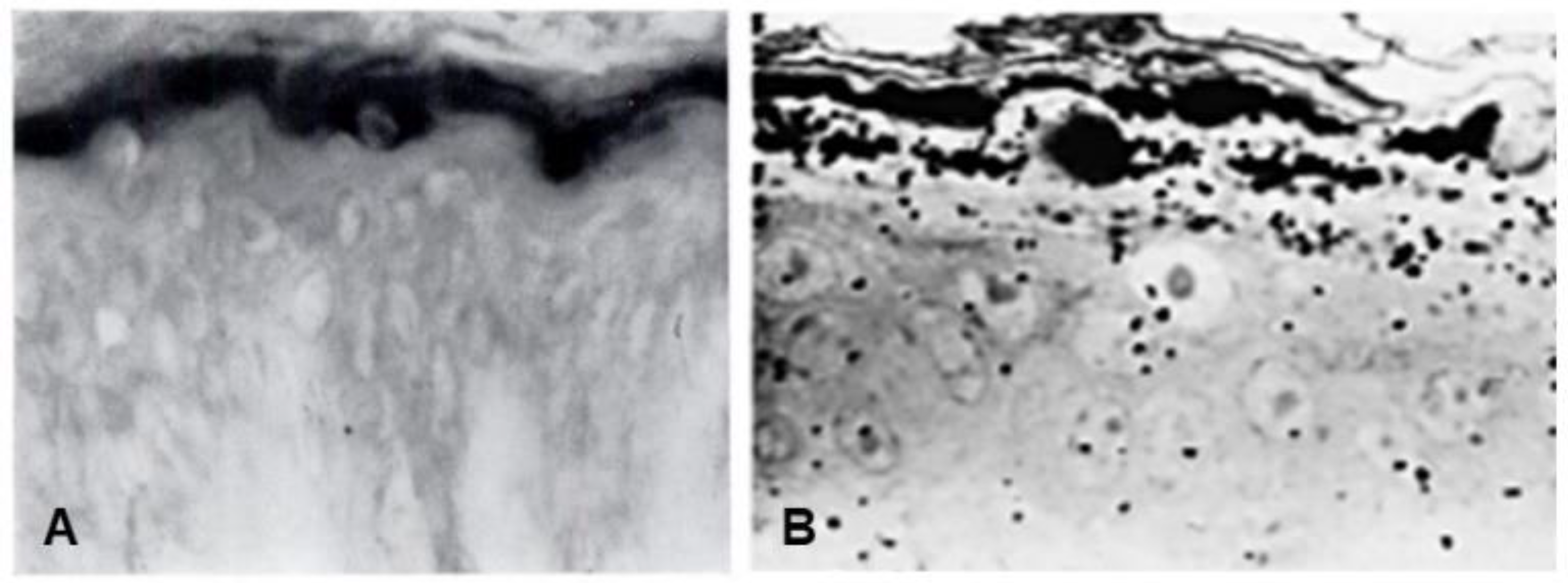
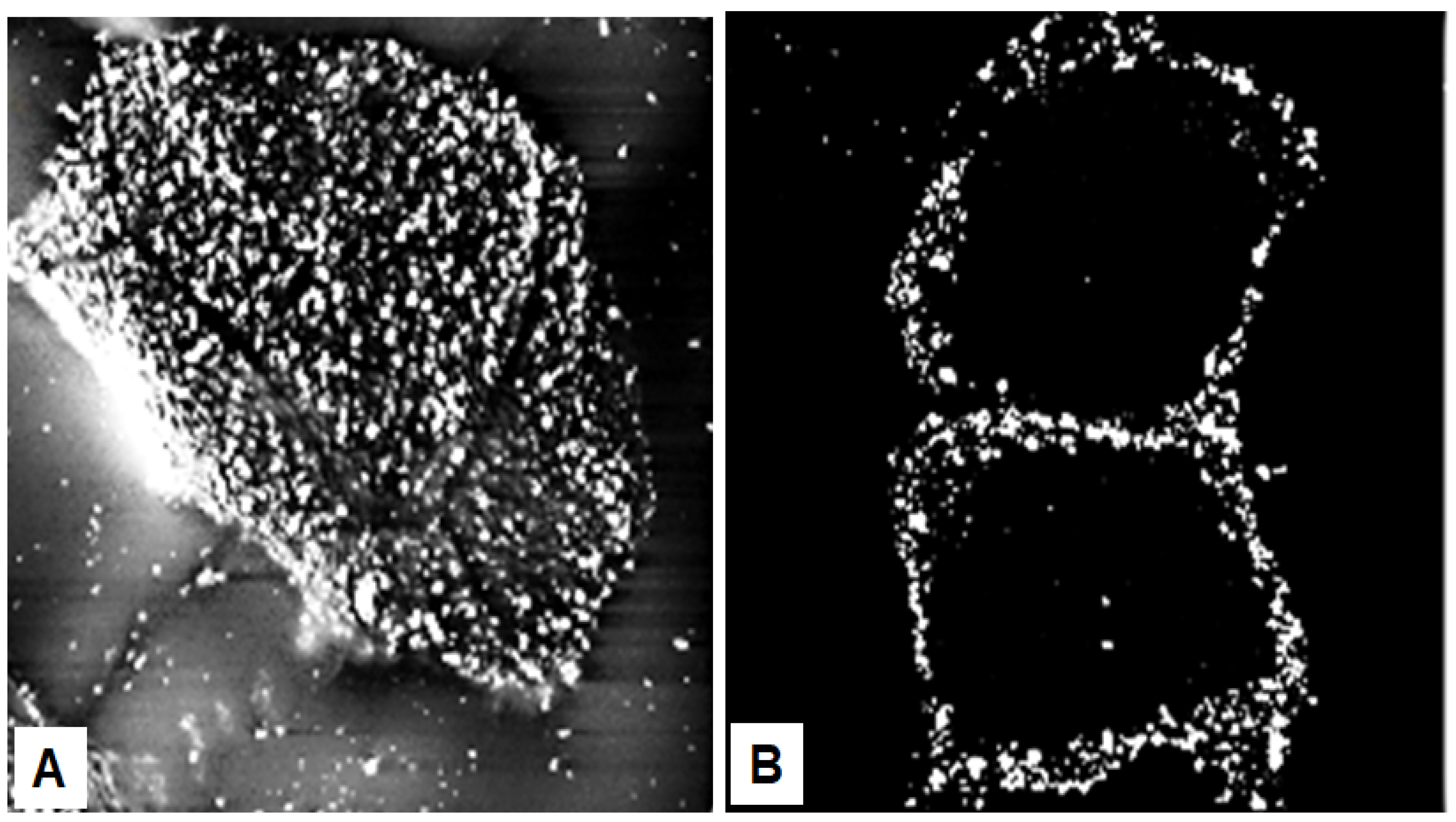
Seminal observations were published in the years following 1960. Reaven and Cox [4][5] traced the accumulation of zinc in the granular layer to chelation by the high density of histidine in the keratohyalin granules [6]. The granules stained an intense red color with diazotized sulfanilic acid under alkaline conditions, a coupling reaction developed by Pauly [7] for histidine (Figure 2A). Kimie Fukuyama, while a visiting scientist in I. A. Bernstein’s laboratory at the University of Michigan, began investigations of the epidermis by the incorporation of radiolabeled nucleotides into DNA and then into RNA by radioautography. She observed that, whereas DNA was synthesized only in the basal layer, incorporation of precursors into RNA occurred throughout the layers below the stratum corneum [8][9], which suggested that proteins were also synthesized in the outer layers. The surprise came when she turned to the incorporation of [3H]-amino acids. As expected, labeled phenylalanine, leucine and methionine were extensively incorporated into the dividing cells of the basal layer but minimally in the outer layers. Conversely, [3H]histidine and [3H]glycine were preferentially incorporated into the granular layer (Figure 2B) [10][11][12]. The possibility of synthesis of a unique protein exclusively in the granular layer while the cells are undergoing terminal differentiation was proposed by Bernstein, who had become interested in the epidermis as a tissue to study the process of differentiation [13]. This hypothesis called for a deeper analysis, which could only be resolved by purification of a protein enriched in histidine and glycine.
2. Discovery of the ‘Histidine-Rich’ Protein
3. The Precursor, Profilaggrin
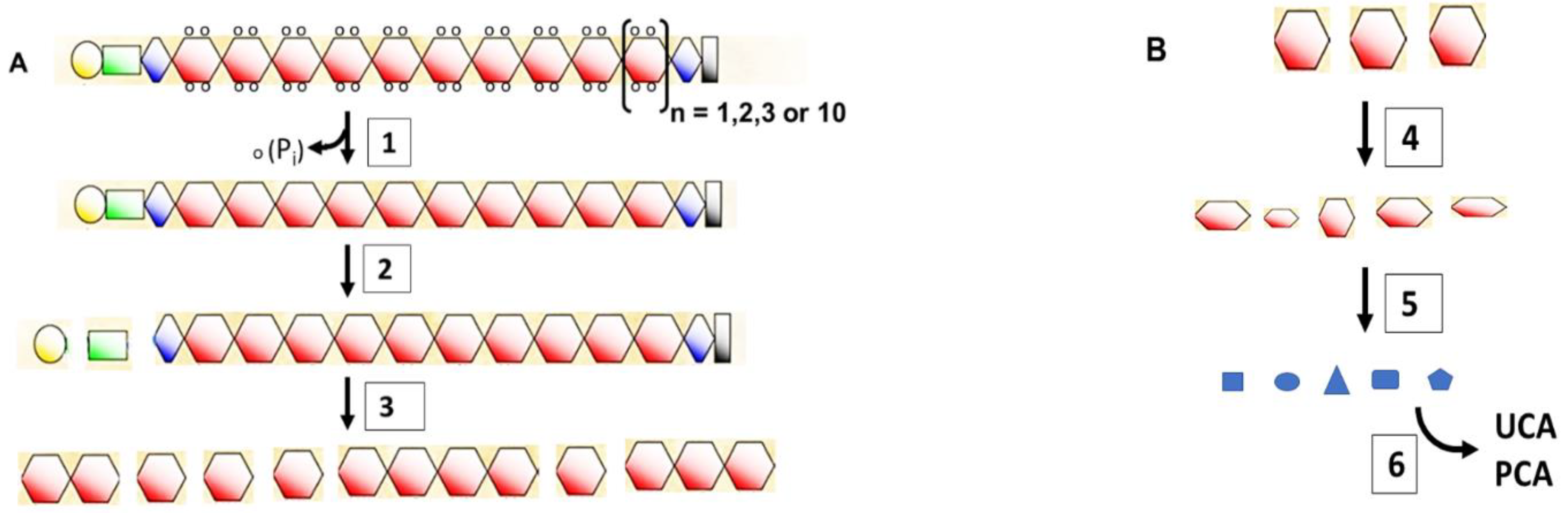
4. The Short Life of Filaggrin
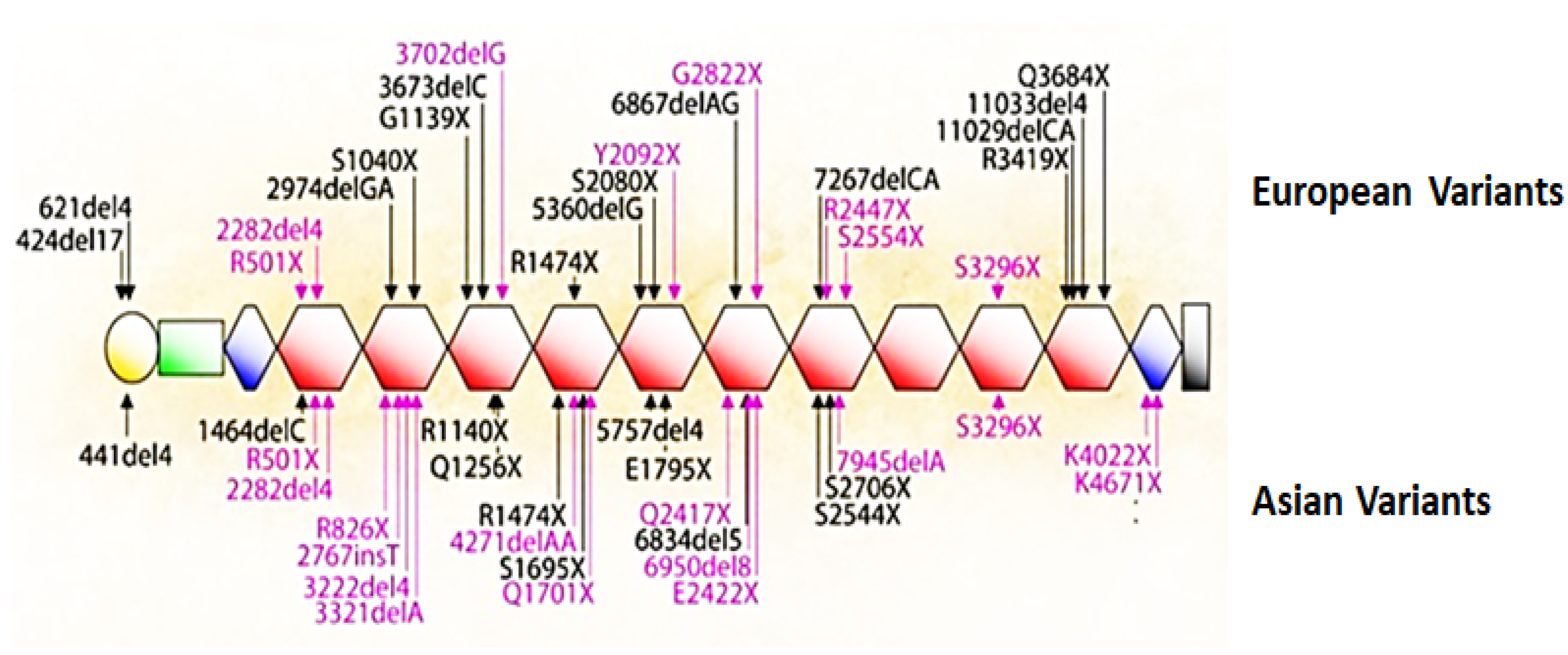
5. Mutations That Cause Loss of Filaggrin
6. Keratinocyte to Corneocyte Transition
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031455
References
- Holbrook, K.A. Biologic structure and function: Perspectives on morphologic approaches to the study of the granular layer keratinocyte. J. Investig. Dermatol. 1989, 92, s84–s104.
- Rothman, S. Keratinization in historical perspective. In The Epidermis; Montagna, W., Lobitz, W.C., Jr., Eds.; Academic Press: New York, NY, USA, 1964; pp. 1–14.
- Burgdorf, W.H.C.; Bickers, D.R. The scientific legacy of Stephen Rothman. J. Investig. Dermatol. 2015, 135, 954–959.
- Reaven, E.P.; Cox, A.J., Jr. The histochemical localization of histidine in the human epidermis and its relationship to zinc binding. J. Histochem. Cytochem. 1963, 11, 782–790.
- Reaven, E.P.; Cox, A.J. Histidine and keratinization. J. Investig. Dermatol. 1965, 45, 422–431.
- Reaven, E.P.; Cox, A.J. Binding of zinc by the transitional layer of the epidermis. J. Investig. Dermatol. 1962, 39, 133–137.
- Pauly, H. Über die Konstitution des Histidins. I. Mitteilung. Z. Physiol. Chem. Hoppe-Seyler 1904, 42, 508–518.
- Fukuyama, K.; Bernstein, I. Autoradiographic Studies of the Incorporation of Thymidine-H3into Deoxyribonucleic Acid in the Skin of Young Rats1. J. Investig. Dermatol. 1961, 36, 321–326.
- Fukuyama, K.; Bernstein, I.A. Site of synthesis of ribonucleic acid in mammalian epidermis. J. Investig. Dermatol. 1963, 41, 47–52.
- Fukuyama, K.; Nakamura, T.; Bernstein, I.A. Differentially localized incorporation of amino acids in relation to epidermal keratinization in the newborn rat. Anat. Rec. 1965, 152, 525–535.
- Fukuyama, K.; Epstein, W.L. Ultrastructural autoradiographic studies of keratohyalin granule formation. J. Investig. Dermatol. 1967, 49, 595–604.
- Fukuyama, K.; Marshburn, I.; Epstein, W.L. Histidine-rich protein in developing rat epidermis. Dev. Biol. 1981, 81, 201–207.
- Bernstein, I.A.; Chakrabarti, S.G.; Kumaroo, K.K.; Sibrack, L.A. Synthesis of protein in the mammalian epidermis. J. Investig. Dermatol. 1970, 55, 291–302.
- Hoober, J.K. The Differential Incorporation of Amino Acids (In Vivo) into Proteins of the Newborn Rat Epidermis. Ph.D. Thesis, University of Michigan, Ann Arbor, MI, USA, 1965.
- Hoober, J.K.; Bernstein, I.A. Studies on the mechanism of the localized incorporation of glycine-H3 in newborn rat epidermis. Fed. Proc. 1963, 22, 238.
- Hoober, J.K.; Bernstein, I.A. Protein synthesis related to epidermal differentiation. Proc. Natl. Acad. Sci. USA 1966, 56, 594–601.
- Gumucio, J.; Feldkamp, C.; Bernstein, I.A. Studies on localization of “histidine-rich” peptide material present in epidermis of the newborn rat. J. Investig. Dermatol. 1967, 49, 545–551.
- Voorhees, J.J.; Siba, M.D.; Chakrabarti, G.; Bernstein, I.A. The metabolism of “histidine-rich” protein in normal and psoriatic keratinization. J. Investig. Dermatol. 1968, 51, 344–354.
- Ball, R.D.; Walker, G.K.; Bernstein, I.A. Histidine-rich proteins as molecular markers of epidermal differentiation. J. Biol. Chem. 1978, 253, 5861–5868.
- Ugel, A.R. Studies on isolated aggregating oligoribonucleoproteins of the epidermis with histochemical and morphological characteristics of keratohyalin. J. Cell Biol. 1971, 49, 405–422.
- Ugel, A.R.; Idler, W. Further characterization of bovine keratohyalin. J. Cell Biol. 1972, 52, 453–464.
- Sibrack, L.A.; Gray, R.; Bernstein, I. Localization of the Histidine-Rich Protein in Keratohyalin: A Morphologic and Macromolecular Marker in Epidermal Differentiation. J. Investig. Dermatol. 1974, 62, 394–405.
- Balmain, A.; Loehren, D.; Alonso, A.; Goerttler, K. Protein synthesis during fetal development of mouse epidermis. II. Biosynthesis of histidine-rich and cystine-rich proteins in vitro and in vivo. Develop. Biol. 1979, 73, 338–344.
- Balmain, A.; Loehren, D.; Fischer, J.; Alonso, A. Protein synthesis during fetal development of mouse epidermis: I. The appearance of “histidine-rich protein”. Dev. Biol. 1977, 60, 442–452.
- Brown, S.J.; McLean, W.I. One Remarkable Molecule: Filaggrin. J. Investig. Dermatol. 2012, 132, 751–762.
- Dale, B.A. Purification and chaaracterization of a basic protein from the stratum corneum of mammalian epidermis. Biochem. Biophys. Acta 1977, 491, 193–204.
- Steinert, P.M.; Cantieri, J.S.; Teller, D.C.; Lonsdale-Eccles, J.D.; Dale, B.A. Characterization of a class of cationic proteins that specifically interact with intermediate filaments. Proc. Natl. Acad. Sci. USA 1981, 78, 4097–4101.
- Harding, C.R.; Scott, I.R. Histidine-rich proteins (filaggrins): Structural and functional heterogeneity during epidermal differentiation. J. Mol. Biol. 1983, 170, 651–673.
- Steinert, P.M.; Roop, D.R. Molecular and cellular biology of intermediate filaments. Ann. Rev. Biochem. 1988, 57, 593–625.
- Fuchs, E.; Green, H. Changes in keratin gene expression during terminal differentiation of the keratinocyte. Cell 1980, 19, 1033–1042.
- McKinley-Grant, L.J.; Idler, W.W.; Bernstein, I.A.; Parry, D.A.; Cannizzaro, L.; Croce, C.M.; Huebner, K.; Lessin, S.R.; Steinert, P.M. Characterization of a cDNA clone encoding human filaggrin and localization of the gene to chromosome region 1q21. Proc. Natl. Acad. Sci. USA 1989, 86, 4848–4852.
- Resing, K.A.; Johnson, R.S.; Walsh, K.A. Characterization of protease processing sites during conversion of rat proflaggrin to filaggrin. Biochemistry 1993, 32, 10036–10045.
- Rothnagel, J.A.; Steinert, P.M. The structure of the gene for mouse filaggrin and a comparison of the repeating units. J. Biol. Chem. 1990, 265, 1862–1865.
- Haydock, P.V.; Dale, B.A. Filaggrin, an Intermediate Filament-Associated Protein: Structural and Functional Implications from the Sequence of a cDNA from Rat. DNA Cell Biol. 1990, 9, 251–261.
- Presland, R.B.; Haydock, P.V.; Fleckman, P.; Nirunsuksiri, W.; Dale, B.A. Characterization of the human epidermal profilaggrin gene. Genomic organization and identification of an S-100-like calcium binding domain at the amino terminus. J. Biol. Chem. 1992, 267, 23772–23781.
- Sandilands, A.; Sutherland, C.; Irvine, A.D.; McLean, W.H.I. Filaggrin in the frontline: Role in skin barrier function and disease. J. Cell Sci. 2009, 122, 1285–1294.
- Ishida-Yamamoto, A.; Senshu, T.; Eady, R.A.J.; Takahashi, H.; Shimizu, H.; Akiyama, M.; Iizukz, H. Sequential reorganization of cornified cell keratin filaments involving filaggrin-mediated compaction and keratin 1 dimination. J. Investig. Dermatol. 2002, 118, 282–287.
- Presland, R.; Kimball, J.R.; Kautsky, M.B.; Lewis, S.P.; Lo, C.Y.; Dale, B.A. Evidence for Specific Proteolytic Cleavage of the N-Terminal Domain of Human Profilaggrin During Epidermal Differentiation. J. Investig. Dermatol. 1997, 108, 170–178.
- Pearton, D.; Nirunsuksiri, W.; Rehemtulla, A.; Lewis, S.P.; Presland, R.; Dale, B.A. Proprotein convertase expression and localization in epidermis: Evidence for multiple roles and substrates. Exp. Dermatol. 2001, 10, 193–203.
- Matsui, T.; Miyamoto, K.; Kubo, A.; Kawasaki, H.; Ebihara, T.; Hata, K.; Tanahashi, S.; Ichinose, S.; Imoto, I.; Inazawa, J.; et al. SASPase regulates stratum corneum hydration through profilaggrin-to-filaggrin processing. EMBO Mol. Med. 2011, 3, 320–333.
- Leyvraz, C.; Charles, R.-P.; Rubera, I.; Guitard, M.; Rolman, S.; Breiden, B.; Sandhoff, K.; Hummler, E. The epidermal barrier function is dependent upon the serine protease CAP1/Prss8. J. Cell Biol. 2005, 170, 487–496.
- List, K.; Szabo, R.; Wertz, P.W.; Segre, J.; Haudenschild, C.C.; Kim, S.-Y.; Bugge, T.H. Loss of proteolytically processed filaggrin caused by epidermal deletion of Matriptase/MT-SP1. J. Cell Biol. 2003, 163, 901–910.
- Hsu, C.-Y.; Henry, J.; Raymond, A.-A.; Méchin, M.-C.; Pendaries, V.; Nassar, D.; Simon, M.; Serre, G.; Paul, C.; Takahara, H.; et al. Deimination of human filaggrin-2 promotes its proteolysis by Calpain1. J. Biol. Chem. 2011, 286, 23222–23233.
- Hoste, E.; Kemperman, P.; Devos, M.; Denecker, G.; Kezic, S.; Yau, N.; Gilbert, B.; Lippens, S.; De Groote, P.; Roelandt, R.; et al. Caspase-14 Is Required for Filaggrin Degradation to Natural Moisturizing Factors in the Skin. J. Investig. Dermatol. 2011, 131, 2233–2241.
- Kamata, Y.; Taniguchi, A.; Yamamoto, M.; Nomura, J.; Ishihara, K.; Takahara, H.; Hibino, T.; Takeda, A. Neutral Cysteine Protease Bleomycin Hydrolase Is Essential for the Breakdown of Deiminated Filaggrin into Amino Acids. J. Biol. Chem. 2009, 284, 12829–12836.
- Hanson, K.M.; Simon, J.D. Epidermal trans-urocanic acid and the UV-A-induced photoaging of the skin. Proc. Natl. Acad. Sci. USA 1998, 95, 10576–10578.
- Gibbs, N.K.; Norval, M. Urocanic Acid in the Skin: A Mixed Blessing? J. Investig. Dermatol. 2011, 131, 14–17.
- Rawlings, A.V.; Harding, C.R. Moisturization and skin barrier function. Dermatol. Ther. 2004, 17, 43–48.
- Kezic, S.; Kemperman, P.M.; Koster, E.S.; de Jongh, C.M.; Thio, H.B.; Campbell, L.E.; Irvine, A.; McLean, W.H.I.; Puppels, G.J.; Caspers, P.J. Loss-of-Function Mutations in the Filaggrin Gene Lead to Reduced Level of Natural Moisturizing Factor in the Stratum Corneum. J. Investig. Dermatol. 2008, 128, 2117–2119.
- Kezic, S.; Novak, N.; Jakasa, I.; Jungersted, J.M.; Simon, M.; Brandner, J.M.; Middelkamp-Hup, M.A.; Weidinger, S. Skin barrier in atopic dermatitis. Front. Biosci. 2014, 19, 542–556.
- De Veer, S.J.; Furio, L.; Harris, J.M.; Hovnanian, A. Proteases: Common culprits in human skin disorders. Trends Mol. Med. 2014, 20, 166–178.
- Smith, F.J.D.; Irvine, A.; Terron-Kwiatkowski, A.; Sandilands, A.; Campbell, L.E.; Zhao, Y.; Liao, H.; Evans, A.T.; Goudie, D.R.; Lewis-Jones, S.; et al. Loss-of-function mutations in the gene encoding filaggrin cause ichthyosis vulgaris. Nat. Genet. 2006, 38, 337–342.
- Irvine, A.; McLean, W.H.I.; Leung, D.Y. Filaggrin Mutations Associated with Skin and Allergic Diseases. N. Engl. J. Med. 2011, 365, 1315–1327.
- McLean, W.H.I. Filaggrin failure—From ichthyosis vulgaris to atopic eczema and beyond. Br. J. Dermatol. 2016, 175 (Suppl. S2), 4–7.
- Salama, R.H.; Rasheed, Z.; Ahmed, A.A.; Bin Saif, G.A.; Elkholy, M.M.; El-Moniem, A.E.A.; Salem, T.; Zedan, K.; Al Robaee, A.A.; Alzolibani, A.A. Missense, silent, non-sense and frame-shift mutations in exon 3 of the filaggrin gene in patients with bronchial asthma, atopic dermatitis, allergic rhinitis and mixed atopy. Nucleosides Nucleotides Nucleic Acids 2021, 40, 357–367.
- Bin, L.; Malley, C.; Taylor, P.; Boorgula, M.P.; Chavan, S.; Daya, M.; Mathias, M.; Shankar, G.; Rafaels, N.; Vergara, C.; et al. Whole genome sequencing identifies novel genetic mutations in patients with eczema herpeticum. Allergy 2021, 76, 2510–2523.
- Esparza-Gordillo, J.; Matanovic, A.; Marenholz, I.; Bauerfeind, A.; Rohde, K.; Nemat, K.; Lee-Kirsch, M.-A.; Nordenskjöld, M.; Winge, M.C.G.; Keil, T.; et al. Maternal Filaggrin Mutations Increase the Risk of Atopic Dermatitis in Children: An Effect Independent of Mutation Inheritance. PLoS Genet. 2015, 11, e1005076.
- Butler, D.C.; Simpson, E.; Guttman-Yassky, E.; Eichenfield, L.F.; Golant, A.K.; Koo, J.Y.M.; Armstrong, A.W.; Alexis, A.F.; Lio, P.A.; Marson, J.W.; et al. The atopic dermatitis spectrum disorder. Recognizing the clinical heterogeneity in patients with atopic related skin conditions in order to improve therapeutic decision-making and outcomes: An expert panel consensus statement. J. Dermatol. Treat. 2021, 33, 1–3.
- Shaw, T.E.; Currie, G.P.; Koudelka, C.W.; Simpson, E.L. Eczema Prevalence in the United States: Data from the 2003 National Survey of Children’s Health. J. Investig. Dermatol. 2011, 131, 67–73.
- Nutten, S. Atopic Dermatitis: Global Epidemiology and Risk Factors. Ann. Nutr. Metab. 2015, 66 (Suppl. S1), 8–16.
- McAleer, M.A.; Jakasa, I.; Raj, N.; O’Donnell, C.P.F.; Lane, M.E.; Rawlings, A.V.; Irvine, A.D.; Voegeli, R.; McLean, W.H.I.; Kezic, S. Early-life regional and temporal variation in filaggrin-derived moisturizing factor, filaggrin-processing enzyme activity, corneocyte phenotypes and plasmin activity: Implications for atopic dermatitis. Br. J. Dermatol. 2018, 179, 431–441.
- Kasparek, P.; Ileninova, Z.; Zbodakova, O.; Kanchev, I.; Benada, O.; Chalupsky, K.; Sedlacek, R.; Brattsand, M.; Beck, I.M. KLK5 and KLK7 ablation fully rescues letahlity of Netherton syndrome-like phenotype. PLoS Genet. 2017, 13, e1006566.
- Furio, L.; Pampalakis, G.; Michael, I.; Nagy, A.; Sotiropoulou, G.; Hovnanian, A. KLK5 Inactivation Reverses Cutaneous Hallmarks of Netherton Syndrome. PLoS Genet. 2015, 11, e1005389.
- Miyai, M.; Matsumoto, Y.; Yamanishi, H.; Yamamoto-Tanaka, M.; Tsuboi, R.; Hibino, T. Keratinocyte-specific mesotrypsin contributes to the desquamation process via kallikrein activation and LEKT1 degradation. J. Investig. Dermatol. 2014, 134, 1665–1674.
- Briot, A.; Deraison, C.; Lacroix, M.; Bonnart, C.; Robin, A.; Besson, C.; Hovnanian, A.; Dubus, P. Kalikrein 5 induces atopic dermatitis-like lesions through PAR2-mediated thymic stromal lymphopoietin expression in Netherton syndrome. J. Exp. Med. 2009, 206, 1135–1147.
- Kalinin, A.; Marekov, L.N.; Steinert, P.M. Assembly of the epidermal cornified cell envelope. J. Cell Sci. 2001, 114, 3069–3070.
- Candi, E.; Schmidt, R.; Melino, G. The cornified envelope: A model of cell death in the skin. Nat. Rev. Mol. Cell Biol. 2005, 6, 328–340.
- Eckert, R.L.; Green, H. Structure and evolution of the human involucrin gene. Cell 1986, 46, 583–589.
- Eckert, R.L.; Sturniolo, M.T.; Broome, A.-M.; Ruse, M.; Rorke, E.A. Transglutaminase Function in Epidermis. J. Investig. Dermatol. 2005, 124, 481–492.
- Simon, M.; Green, H. The glutamine residues reactive in transglutaminase-catalyzed cross-linking of involucrin. J. Biol. Chem. 1988, 263, 18093–18098.
- Ishitsuka, Y.; Roop, D.R. Loricrin: Past, present, and future. Int. J. Mol. Sci. 2020, 21, 2271.
- Steinert, P.M.; Marekov, L.N. The Proteins Elafin, Filaggrin, Keratin Intermediate Filaments, Loricrin, and Small Proline-rich Proteins 1 and 2 Are Isodipeptide Cross-linked Components of the Human Epidermal Cornified Cell Envelope. J. Biol. Chem. 1995, 270, 17702–17711.
- Matsuki, M.; Yamashita, F.; Ishida-Yamamoto, A.; Yamada, K.; Kinoshita, C.; Fushiki, S.; Yamanishi, K.; Ueda, E.; Tabata, K.; Okabe, M.; et al. Defective stratum corneum and early neonatal death in mice lacking the gene for transaminase 1 (keratinocyte transglutamase). Proc. Natl. Acad. Sci. USA 1998, 95, 1044–1049.
- Liedén, A.; Winge, M.C.G.; Sääf, A.; Kockum, I.; Ekelund, E.; Rodriguez, E.; Fölster-Holst, R.; Franke, A.; Illig, T.; Tengvall-Linder, M.; et al. Genetic Variation in the Epidermal Transglutaminase Genes Is Not Associated with Atopic Dermatitis. PLoS ONE 2012, 7, e49694.
- Su, H.; Luo, Y.; Sun, J.; Liu, X.; Ling, S.; Xu, B.; Zhang, Y.; Liu, J.; Li, W.; Wang, B.; et al. Transglutaminase 3 Promotes Skin Inflammation in Atopic Dermatitis by Activating Monocyte-Derived Dendritic Cells via DC-SIGN. J. Investig. Dermatol. 2020, 140, 370–379.
- Griffin, M.; Casadio, R.; Bergamini, C.M. Transglutaminases: Nature’s biological glues. Biochem. J. 2002, 368, 377–396.
- Chou, C.-Y.; Streets, A.J.; Watson, P.F.; Huang, L.; Verderio, E.A.; Johnson, T.S. A Crucial Sequence for Transglutaminase Type 2 Extracellular Trafficking in Renal Tubular Epithelial Cells Lies in Its N-terminal β-Sandwich Domain. J. Biol. Chem. 2011, 286, 27825–27835.
- Zemskov, E.A.; Mikhailenko, I.; Hsia, R.-C.; Zaritskaya, L.; Belkin, A.M. Unconventional Secretion of Tissue Transglutaminase Involves Phospholipid-Dependent Delivery into Recycling Endosomes. PLoS ONE 2011, 6, e19414.
- Nurminskaya, M.V.; Belkin, A.M. Cellular Functions of Tissue Transglutaminase. Int. Rev. Cell. Mol. Biol. 2012, 294, 1–97.
- Telei, D.; Griffin, M. Tissue transaminase (TG2)—A wound response enzyme. Front. Biosci. 2006, 11, 867–882.
- Stephens, P.; Grenard, P.; Aeschlimann, P.; Langley, M.; Blain, E.; Errington, R.; Kipling, D.; Thomas, D.; Aeschlimann, D. Crosslinking and G-protein functions of transglutaminase 2 contribute differentially to fibroblast wound healing responses. J. Cell Sci. 2004, 117, 3389–3403.
- Haroon, Z.A.; Hettasch, J.M.; Lai, T.-S.; Dewhirst, M.W.; Greenberg, C.S. Tissue transglutaminase is expressed, active, and directly involved in rat dermal wound healing and angiogenesis. FASEB J. 1999, 13, 1787–1795.
- Verderio, E.A.M.; Johnson, T.; Griffin, M. Tissue transglutaminase in normal and abnormal wound healing: Review article. Amino Acids 2004, 26, 387–404.
- Johnson, T.S.; Scholfield, C.I.; Parry, J.; Griffin, M. Induction of tissue transglutaminase by dexamethasone: Its correlation to receptor number and transglutaminase-mediated cell death in a series of malignant hamster fibrosarcomas. Biochem. J. 1998, 331, 105–112.
- Pinkas, D.; Strop, P.; Brunger, A.; Khosla, C. Transglutaminase 2 Undergoes a Large Conformational Change upon Activation. PLoS ONE 2007, 5, e327.
- Mearns, B.; Nanda, N.; Michalieck, J.; Iismaa, S.; Graham, R. Impaired wound healing and altered fibroblast cytoskeletal dynamics in Gh knockout mice. Minerva Biotecnol. 2002, 14, 218.
- Elias, P.M. Epidermal lipids, barrier function, and desquamation. J. Investig. Dermatol. 1983, 80, 544–549.
- Nemes, Z.; Steinert, P.M. Bricks and mortar of the epidermal barrier. Exp. Mol. Med. 1999, 31, 5–19.
- Grubauer, G.; Elias, P.M.; Feingold, K.R. Transepidermal water loss: The signal for recovery of barrier structure and function. J. Lipid Res. 1989, 30, 323–333.
- Elias, P.M.; Wakefield, J.S. Mechanisms of abnormal lamellar body secretion and the dysfunctional skin barrier in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2014, 134, 781–791.e1.
- Caubet, C.; Jonca, N.; Brattsand, M.; Guerrin, M.; Bernard, D.; Schmidt, R.; Egelrud, T.; Simon, M.; Serre, G. Degradation of Corneodesmosome Proteins by Two Serine Proteases of the Kallikrein Family, SCTE/KLK5/hK5 and SCCE/KLK7/hK7. J. Investig. Dermatol. 2004, 122, 1235–1244.
- Igawa, S.; Kishibe, M.; Honma, M.; Murakami, M.; Mizuno, Y.; Suga, Y.; Seishima, M.; Ohguchi, Y.; Akiyama, M.; Hirose, K.; et al. Aberrant distribution patterns of corneodesmosomal components of tape-stripped corneocytes in atopic dermatitis and related skin conditions (ichthyosis vulgaris, Netherton syndrome and peeling skin syndrome type B). J. Dermatol. Sci. 2013, 72, 54–60.
- Simon, M.; Montézin, M.; Guerrin, M.; Durieux, J.-J.; Serre, G. Characterization and Purification of Human Corneodesmosin, an Epidermal Basic Glycoprotein Associated with Corneocyte-specific Modified Desmosomes. J. Biol. Chem. 1997, 272, 31770–31776.
- Guerrin, M.; Simon, M.; Montézin, M.; Haftek, M.; Vincent, C.; Serre, G. Expression Cloning of Human Corneodesmosin Proves Its Identity with the Product of the S Gene and Allows Improved Characterization of Its Processing during Keratinocyte Differentiation. J. Biol. Chem. 1998, 273, 22640–22647.
- Riethmuller, C.; McAleer, M.A.; Koppes, S.A.; Abdayem, R.; Franz, J.; Haftek, M.; Campbell, L.E.; MacCallum, S.F.; McLean, W.I.; Irvine, A.D.; et al. Filaggrin breakdown products determine corneocyte conformation in patients with atopic dermatitis. J. Allergy Clin. Immunol. 2015, 136, 1573–1580.e2.