Alphaviruses are members of the Togaviridae family that are mainly transmitted by arthropods such as mosquitoes. In the last decades, several alphaviruses have re-emerged causing outbreaks worldwide. Infections with the Old World alphaviruses (e.g. CHIKV, RRV) are primarily associated with polyarthritis and myalgia that can persist for months to years. On the other hand, New World alphaviruses such as VEEV cause mainly neurological disease. Despite the worldwide (re-)emergence of these viruses, there are no antivirals or vaccines available for the treatment or prevention of infections with alphaviruses. It is therefore of utmost importance to develop antiviral strategies against these viruses. We here provide an overview of the reported antiviral strategies against arthritogenic alphaviruses. In addition, we highlight the future perspectives for the development and the proper use of such antivirals.
- arbovirus
- alphavirus
- chikungunya
- antivirals
- capping
- protease
- replication
1. Introduction
Alphaviruses are a group of human and animal viruses that belong to the family Togaviridae. Alphaviruses are mainly transmitted by the bite of hematophagous arthropods (e.g., ticks and mosquitoes) [1]. Classically, alphaviruses are divided into the New World and Old World alphaviruses based on their historical geographical abundance. New World alphaviruses cause encephalitic diseases and include the Venezuelan and Western equine encephalitis viruses (VEEV and WEEV) [2]. Infections with Old World alphaviruses, such as the chikungunya virus (CHIKV) and Ross River virus (RRV), mainly result in rheumatic disease that causes debilitating pain in the joints [2]. The symptoms of acute disease caused by these viruses include fever, bilateral symmetrical arthritis, and sometimes skin rash. Although acute infections by arthritogenic alphaviruses are self-limiting, several patients suffer from a chronic polyarthritis that can severely incapacitate the patient for weeks and even up to several years after the acute stage [3]. Recent outbreaks of arthritogenic alphaviruses such as CHIKV have also been associated with neurological manifestations, e.g., myelopathy, Guillain-Barré syndrome, and meningoencephalitis, especially in elderly patients with comorbidities and neonates [4].
Several arthritogenic alphaviruses have (re-)emerged worldwide and have become a major public health threat. However, there is no approved antiviral drug or vaccine for the treatment or prevention of these viral infections. The current treatment depends on symptomatic relief using analgesics, antipyretics, nonsteroidal anti-inflammatory drugs, and, in severe cases, methotrexate [2].
2. Antiviral Strategies
In theory, all steps in the alphavirus replication cycle can be potential targets for antiviral drug development. These steps involve an interplay between viral proteins and host factors. The major advantage of host-targeting antivirals is the potential broad-spectrum activity against more than one alphavirus and the lower possibility that the virus will develop resistance. However, caution must be taken to avoid (serious) side effects when targeting a host factor. We here reviewed the reported antiviral strategies against arthritogenic alphaviruses replication.
2.1. Virus-Targeting Inhibitors
2.1.1. Early-Stage Inhibitors
To date, the entry receptors for arthritogenic alphaviruses are not fully elucidated. Interestingly, the adhesion molecule Mxra8 has been recently identified as an entry receptor for several arthritogenic alphaviruses including CHIKV, ONNV, RRV, and MAYV [13]. Treatment of different cell lines with a Mxra8–Fc fusion protein or anti-Mxra8 monoclonal antibody proved to inhibit CHIKV infection. In addition, blocking Mxra8 with both molecules reduced CHIKV and ONNV infection and disease symptoms in C57BL/6 mice [13].
Another possible target for blocking viral entry is prohibitin. Prohibitin-1 is a signaling protein that was previously identified as a receptor for CHIKV in mammalian cells [14]. Targeting prohibitin-1 with synthetic flavaglines derivatives (Figure 1) inhibited the in vitro CHIKV replication and reduced the colocalization of prohibitin-1 and the CHIKV E2 glycoprotein, suggesting an effect on CHIKV binding to this receptor [15].
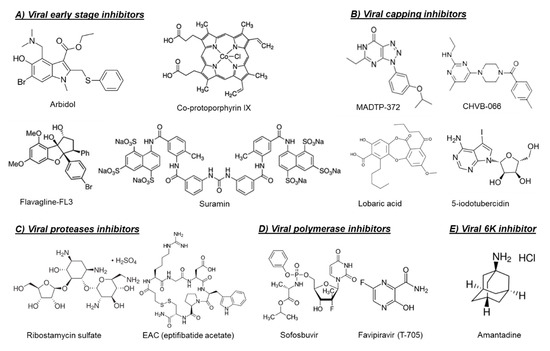
The broad-spectrum antiviral drug arbidol (Figure 1) has also been reported as an early-stage inhibitor of CHIKV replication in vitro [16]. An arbidol-resistant CHIKV variant was identified to carry a glycine to an arginine (G407R) mutation in the viral E2 glycoprotein, which is the protein involved in the viral binding to host receptors [16]. Suramin, an anti-trypanosomiasis drug (Figure 1), has also been reported to inhibit the replication of different CHIKV isolates and related alphaviruses in vitro and in vivo [17,18,19]. Suramin proved to interact directly with the viral particles of SFV and CHIKV and hence prevented viral attachment to the host cells [20]. Moreover, suramin showed the ability to interfere with the conformational changes of viral envelope glycoproteins required for the fusion step [20].
2.1.2. Viral Capping Inhibitors
2.1.3. Viral Protease Inhibitors
2.1.4. Viral RNA-Dependent-RNA Polymerase Inhibitors
2.1.5. Viral 6K Inhibitors
2.1.6. Virucidal Compounds
2.2. Host-Targeting Inhibitors
2.2.1. Endosomal Fusion Inhibitors
2.2.2. Lipid Pathways Inhibitors
2.2.3. Protein Synthesis Inhibitors
2.2.4. Nucleotide Depleting Compounds
2.2.5. Cellular Kinase Modulators
2.2.6. Cellular Chloride Channels Inhibitors
2.2.7. Cellular Furin Inhibitors
2.2.8. Sodium-Potassium ATPase Inhibitors
2.2.9. Serotonin Receptors Modulators
2.2.10. Immunomodulators
3. Perspectives
The worldwide re-emergence of arthritogenic alphaviruses and the high morbidity rate associated with their infections underline the need for potent and safe antiviral drugs against these viruses. Efficacious antiviral drugs, if administered in time, could reduce the severity of the disease symptoms during the acute infection by lowering the viral loads in the infected patient. Moreover, reducing viremia in infected patients using antivirals may indirectly limit virus transmission by mosquitoes and hence might reduce the chance for massive epidemics [99]. Since the severity of symptoms during the acute phase of infection could increase the chance for alphavirus-induced chronic polyarthritis [100], the use of antivirals during the acute infection may decrease the likelihood to develop chronic symptoms.
Currently, approved antiviral drugs are available for the treatment of only a limited number of viruses such as HIV, HBV, HCV, influenza, and herpes viruses. By investing sufficient time and efforts, it could also be possible to develop safe and potent antivirals for the treatment and/or prophylaxis of arthritogenic alphavirus infections. As discussed in this review, several molecules with in vitro anti-alphavirus activity have been reported, but most of these are still in the early stages of preclinical development. A next important and indispensable step is the evaluation of in vivo infection models. Only few of the reported molecules have been tested in alphavirus infection models so far. Infection models in small animals are available for several alphaviruses [1], but the existing models have limitations since they do not recapitulate all the key aspects of alphavirus disease in humans.
The development of specific antiviral drugs against each arthritogenic alphavirus separately will not be economically viable. Therefore, pan-alphavirus inhibitors will be the prime strategy to cope with this challenge. Since the alphavirus nsP2 (protease) and nsP4 (viral polymerase) proteins have conserved catalytic domains, both are considered potential targets for development of broad-spectrum antivirals for arthritogenic alphaviruses. Another promising target is Mxra8, as it functions as an entry receptor for several arthritogenic alphaviruses [13]. Designing molecules that can specifically block this receptor may therefore be a broad-spectrum strategy to control infections with various arthritogenic alphaviruses. Besides virus-specific antivirals that target a conserved alphavirus target, host-targeting antivirals have the potential to be broad-spectrum as well. The heparan sulfate mimetic pentosan polysulfate for example has already advanced to phase II clinical studies for RRV and could be considered for evaluation as a treatment for other arthritogenic alphaviruses.
Another beneficial strategy to control emerging viral infections is the repurposing of drugs that have been approved for other diseases. Because repurposed drugs have already been intensively studied in patients and thus the safety profile is well known, the clinical evaluation of such drugs could be fast-tracked during viral epidemics. Furthermore, production strategies for these drugs have been implemented. However, repurposed drugs cannot be expected to be highly potent inhibitors of alphaviruses, as these were not developed specifically against this particular virus. Examples of approved drugs that would be of interest to evaluate in clinical trials during epidemics of arthritogenic alphaviruses include sofosbuvir, favipiravir, orlistat, and tilorone, since these drugs showed promising antiviral activities, not only in cell culture, but also in vivo animal models.
Another challenge for alphavirus antiviral drug development is how such an antiviral drug could be used practically. The viremia of alphaviruses is short (<1 week). Therefore, antiviral therapy that targets virus replication should be initiated soon after the start of the infection to be efficacious. This requires an early diagnosis, but this is currently difficult. The therapeutic use of antiviral drugs could therefore be complicated. Pre-exposure prophylaxis of household members of an infected patient has been proposed as a useful strategy, since the probability of arthritogenic alphavirus transmission such as CHIKV has been reported to be up to 12% between household members [101]. Another potential application of antivirals against arthritogenic alphaviruses is prophylaxis of travelers before visiting an endemic area of a certain alphavirus. To be used as a prophylactic, an antiviral drug must be very safe and preferentially without any side effects. Prophylactic use of anti-alphavirus drugs might thus be challenging as well. To cope with the health burden and threat of (re-)emerging arthritogenic alphaviruses, the development of pan-alphavirus inhibitors will be important. More research is required to validate different approaches to obtain such pan-alphavirus inhibitors. Furthermore, a better understanding of the alphavirus life cycle and of alphavirus-induced disease is essential to provide insights that will aid to the development of broad-spectrum antiviral strategies for (arthritogenic) alphaviruses.
This entry is adapted from the peer-reviewed paper 10.3390/microorganisms8091365