Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
S-15176 difumarate salt, a derivative of the anti-ischemic metabolic drug trimetazidine, has been intensively studied for its impact on cellular metabolism in animal models of ischemia-reperfusion injury of the liver, heart, spinal cord, and other organs. Despite evidence of some reduction in oxidative damage to cells, the results of therapy with S-15176 have been mostly disappointing, possibly because of the lack of data on its underlying mechanisms.
- S-15176
- mitochondria
- mitochondrial membrane potential
- mitochondrial respiration
- electron transport chain
- mitochondrial membrane permeabilization
1. Introduction
Trimetazidine (TMZ), the derivative of piperazine, is an anti-ischemic metabolic agent of a potent class of drugs called partial inhibitors of fatty acid oxidation (pFOX). Meta-analyses of clinical studies have proved the therapeutic effect of TMZ against stable angina. Nowadays, TMZ is included in the guidelines of the European Society of Cardiology for the management of stable angina pectoris and stable coronary artery disease [1,2,3]. Some studies have demonstrated that TMZ has a cytoprotective effect in models of ischemic damage to the kidney and liver [4,5], as well as in several metabolic pathologies, including diabetes mellitus [6,7,8]. The beneficial effect of TMZ on ischemic tissues is mainly attributed to its inhibitory action on the long-chain 3-ketoacyl-coenzyme A thiolase, resulting in inhibition of the beta-oxidation pathway of free fatty acids [9,10,11]. TMZ can also increase the activity of pyruvate dehydrogenase, which is the rate-limiting enzyme of glucose oxidation under aerobic conditions [10,11,12]. This, by switching cellular metabolism from the oxidation of free fatty acids towards the utilization of carbohydrates, would improve the efficiency of ATP synthesis for a given amount of oxygen molecules consumed, thereby maintaining the energy supply in hypoxia [12]. Taken together, TMZ can shift the energy substrate metabolism, enhancing glucose metabolism and decreasing oxygen consumption. This may be accompanied by decreased ROS production, a limited increase in intracellular acidosis, and reduced accumulation of cytosolic calcium. All of these events are related to the mitochondria, which are likely to have several targets for this drug. Recent studies suggest that TMZ inhibits the opening of the mitochondrial permeability transition (MPT) pore, contributing to protection against dysfunction of mitochondria and cell death in response to ischemia [13,14,15].
S-15176 (N-{(3,5-di-tert-butyl4-hydroxy-1-thiophenyl)}-3-propyl-N′-(2,3,4-trimethoxybenzyl) piperazine) difumarate salt is a derivative of TMZ (Figure S1), but its therapeutic efficacy and mechanisms of action have not yet been fully understood. The drug can act on several targets in the mitochondria. In particular, S-15176 has been reported to inhibit the rate-limiting enzyme of the beta-oxidation cycle carnitine palmitoyltransferase I (CPT-1) [16], suppress the formation of the MPT pore and the cyclosporin A-insensitive mitochondrial pore, and prevent free radical-induced toxicity [17,18]. These effects are suggested to underlie the protective action of S-15176 against mitochondrial and cellular dysfunction in ischemia–reperfusion injury of liver and myocardium tissues, traumatic spinal cord injury, and experimental diabetes mellitus [19,20,21,22,23].
Some studies on isolated mitochondria revealed that, in addition to the above effects, S-15176 could induce mitochondrial uncoupling and reverse the activity of the ATP synthase, leading to the hydrolysis of ATP molecules [17,24]. However, the mechanism of these processes remains unknown. It was suggested that the S-15176-induced decrease in the mitochondrial membrane potential proceed through a disruption of the function of the respiratory chain complexes. One can assume that treatment with S-15176 may lead to mitochondrial dysfunction, impaired energy metabolism, ion dyshomeostasis, and, ultimately, cell death in tissues that are most vulnerable to the toxic action of chemicals or exposed to higher doses of the agent. Therefore, a study of the role of the mitochondrial membrane potential and mitochondrial enzyme complexes of the respiratory chain in S-15176-induced toxicity is needed, as well as a study of the involvement of mitochondrial ROS production and permeability transition, which could potentially provide possibilities for interventions on the adverse effects caused by this agent.
Based on these considerations, we used rat thymocytes and isolated rat liver mitochondria as model objects to examine the effect of S-15176 at different concentrations on the mitochondrial membrane potential and mitochondrial respiratory capacities when using different combinations of respiratory substrates. To determine which complex(es) of the electron transfer system is (are) affected by S-15176, we estimated the enzymatic activity of complexes I-IV in the presence of this agent. We then evaluated its effects on the generation of mitochondrial ROS and Ca2+-induced permeability transition as possible mechanisms of the drug-associated mitotoxicity. Our results indicated that acute exposure to S-15176 resulted in a higher dissipation of the membrane potential of the organelles energized with succinate or malate/glutamate, but not ascorbate/TMPD. In addition, S-15176 difumarate salt inhibited complex I- and complex II-linked ADP-stimulated respiration but had no effect on CIV-linked ADP-stimulated respiration, which could be due to its direct inhibitory action on enzyme complex III of the mitochondrial electron transfer system. We also found that low doses of S-15176 diminished the production of H2O2 and increased the calcium retention capacity index, while at concentrations above 30 μM, S-15176 contributed to mitochondrial membrane permeabilization.
2. S-15176 Difumarate Salt Promotes Mitochondrial Depolarization in Rat Thymocytes
Increasing evidence suggests that S-15176 difumarate salt acts on several targets in the mitochondria [16,17,18,24]. In this study, we evaluated whether this drug affects the key indicators of mitochondrial activity. The mitochondrial membrane potential is known to be the main index of mitochondrial function since it reflects the processes of electron transfer and oxidative phosphorylation. Our results demonstrated that S-15176 difumarate salt could initiate mitochondrial depolarization in rat thymocytes. As shown in Figure 1, preincubation of rat thymocytes with S-15176 at concentrations of 10 and 30 µM for 30 min resulted in a decline in the mitochondrial membrane potential in a dose-dependent manner. Importantly, the addition of S-15176 at a concentration of 30 µM led to complete mitochondrial depolarization in the whole cell population.
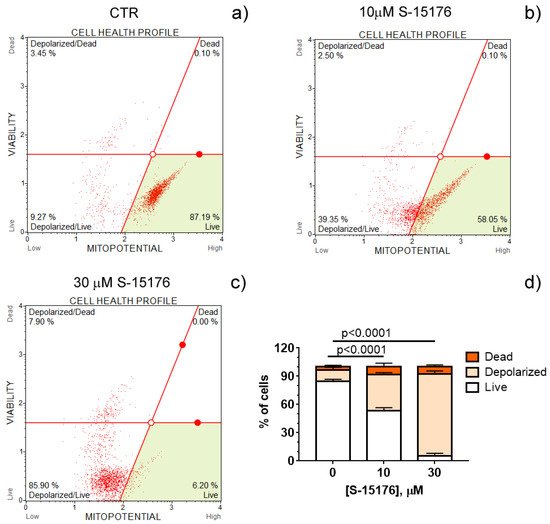
Figure 1. S-15176 difumarate salt induces the depolarization of the mitochondrial membrane in rat thymocytes. Mitochondrial membrane potential (MitoPotential) was assessed with the Muse Cell Analyzer using the Muse MitoPotential kit. Cells were treated with 0 μM S-15176 (a), 10 μM S-15176 (b), or 30 μM S-15176 (c) for 30 min. Typical profile plots are presented. Panel (d) shows the ratio (%) of living, depolarized, and dead rat thymocytes in the presence of S-15176 at different concentrations. In control experiments (CTR, 0 µM S-15176), an equivalent volume of the solvent (0.1% DMSO) was used. Data represent the mean ± SEM (n = 4).
3. S-15176 Inhibits ADP- and 2,4-Dinitrophenol- Stimulated Mitochondrial Respiration Due to Suppression of the Enzymatic Activity of the Respiratory Complex III
To elucidate the mechanism of mitochondrial depolarization caused by S-15176, we investigated the effect of the agent on the efficiency of oxidative phosphorylation (OXPHOS) and the enzymatic activity of the individual OXPHOS complexes I–IV in isolated mitochondria.
The influence of S-15176 on the mitochondrial bioenergetics was assessed by the rate of oxygen consumption by rat liver mitochondria in the main metabolic states using different combinations of respiration substrates. Table 1 demonstrates the effect of S-15176 on the respiration rates of mitochondria oxidizing the substrates of complex I (2.5 mM glutamate and 2.5 mM malate) or complex II (5 mM succinate in the presence of 1 μM rotenone). One can see that when using both of these combinations of substrates, S-15176 difumarate salt stimulated mitochondrial respiration under resting conditions (States 4 and 2) in a dose-dependent manner (Table 1). Moreover, S-15176 dose-dependently suppressed the rates of mitochondrial respiration in the presence of ADP (State 3) or the uncoupler 2,4-dinitrophenol (DNP) (State 3UDNP). In parallel, the respiratory control ratio (RCR) (State 3/State 4), which is directly related to the OXPHOS coupling efficiency, was reduced by 2.1 and 1.8 times in the presence of 30 µM S-15176 when using the substrates of complex I and complex II, respectively. Furthermore, the successive addition of 10 μM S-15176 pulses to the mitochondrial suspension resulted in a gradual decrease in the membrane potential of mitochondria energized by complex I- or complex II-linked respiratory substrates (Figure 2). The latter is consistent with the literature data that S-15176 can display an uncoupling activity in rat liver mitochondria [24].
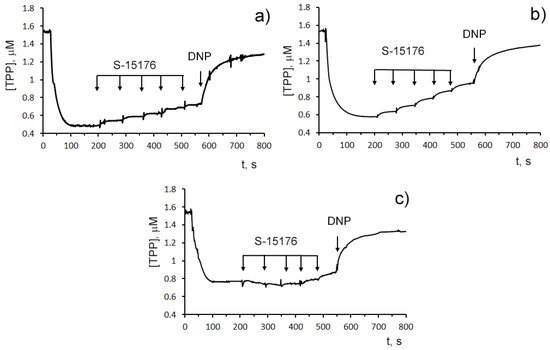
Figure 2. Sequential addition of 10 μM S-15176 gradually decreases the membrane potential of isolated rat liver mitochondria energized by the complex I substrates (2.5 mM potassium glutamate + 2.5 mM malate) (a), or the complex II substrate (5 mM potassium succinate in the presence of 1 µM rotenone) (b), but not by the complex IV substrates (5 mM ascorbic acid + 0.2 mM TMPD) (c). Mitochondrial membrane potential was estimated by the distribution of tetraphenylphosphonium bromide (TPP+) with an ion-sensitive electrode. Additions: 10 µM S-15176 (five pulse additions), 50 µM DNP. Typical traces are shown (n = 6).
Table 1. Effects of S-15176 difumarate salt on the respiration of rat liver mitochondria.
| S-15176, μM | V Respiration, nmol O2 × min−1 × mg−1 Protein | ||||
|---|---|---|---|---|---|
| State 2 | State 3 | State 4 | State 3UDNP | RCR | |
| Glutamate + malate | |||||
| 0 | 3.1 ± 0.2 | 21.5 ± 1.1 | 3.5 ± 0.1 | 21.8 ± 1.0 | 5.9 ± 0.1 |
| 10 | 4.0 ± 0.3 * | 18.1 ± 0.8 * | 3.9 ± 0.2 | 17.9 ± 0.9 * | 4.7 ± 0.1 *** |
| 30 | 4.5 ± 0.3 ** | 16.0 ± 0.2 ** | 5.9 ± 0.1 ** | 15.9 ± 0.2 ** | 2.7 ± 0.1 *** |
| Succinate | |||||
| 0 | 6.0 ± 0.3 | 32.8 ± 0.6 | 6.9 ± 0.2 | 47.2 ± 1.2 | 4.8 ± 0.1 |
| 10 | 7.7 ± 0.2 ** | 30.7 ± 0.6 * | 8.4 ± 0.3 ** | 44.1 ± 0.8 | 3.7 ± 0.2 ** |
| 30 | 8.9 ± 0.2 ** | 29.3 ± 0.9 * | 10.8 ± 0.3 ** | 40.9 ± 1.4 * | 2.7 ± 0.1 *** |
| Ascorbate + TMPD | |||||
| 0 | 25.4 ± 0.6 | 34.9 ± 1.0 | 23.7 ± 0.4 | 38.9 ± 1.3 | 1.5 ± 0.1 |
| 10 | 27.1 ± 0.8 | 35.3 ± 0.8 | 25.0 ± 0.5 | 37.7 ± 0.9 | 1.4 ± 0.1 |
| 30 | 29.0 ± 0.5 ** | 35.7 ± 0.5 | 26.7 ± 0.3 * | 36.4 ± 1.1 | 1.3 ± 0.1 * |
Oxygen consumption of mitochondria was fueled by 2.5 mM glutamate and 2.5 mM malate, 5 mM succinate (in the presence of 1 μM rotenone), and 5 mM ascorbate + 0.2 mM TMPD. Data represent the mean ±SEM (n = 5). * p < 0.05, ** p < 0.01, *** p < 0.001—differences between the control (with 0.1% DMSO, 0 µM S-15176) and the experiment (with 10 or 30 µM S-15176) were statistically significant.
By contrast, when the substrates needed for the complex IV activity (ascorbate and TMPD) were used, S-15176 at concentrations of 10 and 30 µM had no effect on the rate of oxygen consumption by mitochondria in States 3 and 3UDNP (Table 1). In this case, the addition of 30 µM S-15176 led to a decrease in the RCR by only 9%, which was due to a slight stimulation of the resting state respiration (State 4) with the agent. Importantly, the S-15176-induced decline in the membrane potential of rat liver mitochondria incubated with ascorbate + TMPD was observed only at high concentrations of this agent (above 40 µM), and it was significantly less pronounced compared with that when using malate + glutamate or succinate as substrates (Figure 2). It should be noted that similar results were obtained when using the fluorescent dye safranin O as an indicator of the mitochondrial membrane potential (Figure S2). These findings suggest that, in addition to the proton conductance activity, S-15176 has the ability to directly impair certain components of the mitochondrial electron transfer system.
Indeed, a spectrophotometrical assay of the activity of OXPHOS enzymes revealed that 30 μM S-15176 decreased the activity of complex III by 1.6 times but did not affect complexes I, II, and IV in isolated mitochondria (Table 2).
Table 2. Effect of S-15176 difumarate salt (30 μM) on the activity of complexes (CI-IV) of the mitochondrial respiratory chain.
| Values in % of Activity Compared with the Control (100%) | |||
|---|---|---|---|
| CI | CII | CIII | CIV |
| 102.7 ± 1.9 | 99.4 ± 3.2 | 63.3 ± 1.4 * | 105.1 ± 3.7 |
In the absence of S-15176 (control, 0.1% DMSO), the activities of complexes I, II, III, and IV were 385 ± 5, 435 ± 4, 679 ± 9, 467 ± 17 nmol*min−1*mg−1, respectively. The activity values in the absence of S-15176 were taken as 100%. Data represent the mean ±SEM (n = 3). * Differences between the control (0.1% DMSO) and the experiment (30 µM S-15176) were statistically significant (p < 0.05).
4. S-15176 Inhibits H2O2 Production by Rat Liver Mitochondria
Next, we measured the effect of S-15176 on the rate of H2O2 generation in rat liver mitochondria (Figure 3). Our results showed that at concentrations of 10 and 30 µM, S-15176 significantly inhibited the rate of H2O2 production by the mitochondria. However, this effect was canceled with a further increase in the concentration of the drug. The addition of 50 µM S-15176 to the mitochondria returned the rate of H2O2 formation to the control level.
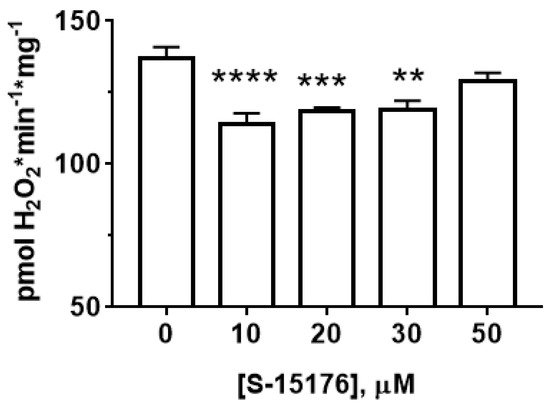
Figure 3. Effect of S-15176 on the rate of H2O2 production by rat liver mitochondria. Data represent the mean ± SEM (n = 5). ** p < 0.01, *** p < 0.005, and **** p < 0.001.
5. S-15176 at Low Concentrations Suppress the Opening of the Ca2+-Dependent MPT Pore and, at High Concentrations, the Agent Itself Contributes to the Permeabilization of the Mitochondrial Membrane
Mitochondrial Ca2+ overload is one of the key mechanisms involved in the opening of the MPT pore and mitochondrial damage [29]. Some studies demonstrated that S-15176 could inhibit the formation of the Ca2+-dependent MPT pore in mitochondria. We observed that 10 μM S-15176 suppressed the high-amplitude swelling of rat liver mitochondria induced by 50 μM Ca2+ in the presence of 1 mM Pi (Figure 4a). Alternatively, 30 μM S-15176 by itself induced a decrease in the optical density of the mitochondrial suspension (Figure 4b), indicating the beginning of the swelling process. It should be noted that the amplitude of mitochondrial swelling induced by S-15176 was less than that after the MPT induction. No further increase in the amplitude of S-15176-induced mitochondrial swelling was observed when using the concentrations of S-15176 above 30 μM. It is important to note that the addition of the selective inhibitor of the MPT pore opening cyclosporin A did not affect the S-15176-induced swelling of mitochondria (Figure 4c), supporting the conclusion that S-15176 in high concentrations could promote mitochondrial membrane permeabilization through an MPT-independent mechanism.
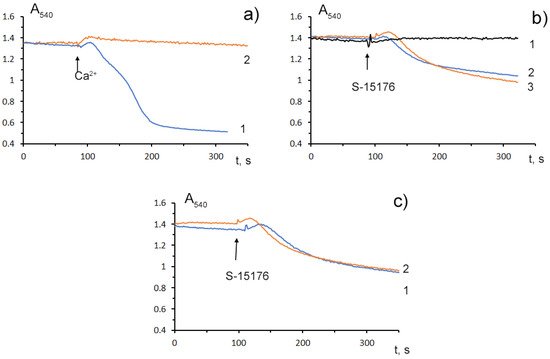
Figure 4. Swelling assay of rat liver mitochondria (0.35 mg/mL) treated with S-15176 difumarate salt; (a) Preincubation of mitochondria with 10 μM S-15176 for 1 min (2) blocks mitochondrial swelling induced by 50 μM Ca2+ in the presence of 1 mM Pi (1); (b) S-15176 by itself induces mitochondrial swelling when its concentration is increased from 10 µM (1) to 30 µM (2) or 50 µM (3); (c) Pretreatment of rat liver mitochondria with 1 μM cyclosporin A (2) does not affect mitochondrial swelling induced by 30 μM S-15176 (1). Typical traces are shown (n = 5).
To determine whether S-15176 can reduce the susceptibility of mitochondria to the formation of the classical MPT pore, we next assessed the calcium retention capacity index, which reflects the maximum overload with Ca2+ that occurs immediately before the MTP pore formation. Our results demonstrated that 10 µM S-15176 increased this indicator, while 50 μM S-15176 stimulated the Ca2+-induced MPT pore opening (Figure 5). These observations allow us to conclude that S-15176 has a dual effect on mitochondrial Ca2+ overload depending on the concentration used.
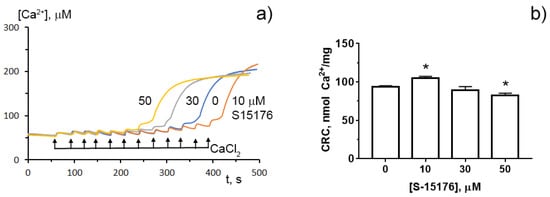
Figure 5. Typical changes in the external (Ca2+) upon the successive addition of 10 μM CaCl2 pulses to the suspension of rat liver mitochondria in the presence of S-15176 (10, 30, and 50 µM) or 0.1% DMSO (0 µM S-15176); (a) The action of S-15176 difumarate salt on the calcium retention capacity index of rat liver mitochondria; (b) The index reflects maximum Ca2+ overload of mitochondria that occurs immediately before the MTP pore opening. Data represent the mean ± SEM (n = 5). * p < 0.05.
6. Conclusions
S-15176 difumarate salt can interact with mitochondria and affect their function through the regulation of several molecular targets. As shown in the current investigation, despite the positive effect of low doses of S-15176 on some mitochondrial functions, acute exposure to this drug in concentrations over 30 µM can be toxic to the mitochondria and cells. Our in vitro studies showed that 30 µM S-15176 could decrease complex I and II-linked respiration due to direct inhibition of the activity of the mitochondrial respiratory complex III coenzyme Q:cytochrome c-oxidoreductase and promote cyclosporin A-insensitive mitochondrial swelling. These effects of S-15176, along with its known protonophoric activity, are likely to underlie the decline in the membrane potential, the driving force behind ATP production, which was observed in both rat thymocytes and isolated liver mitochondria. Altogether, these findings suggest that S-15176 at tissue concentrations reached in animals can cause mitochondrial dysfunction through similar mechanisms in organs that are most vulnerable to chemical toxicity or exposed to higher concentrations of the drug.
This entry is adapted from the peer-reviewed paper 10.3390/biology11030380
This entry is offline, you can click here to edit this entry!