2. Molecular Pathology of Vascular Damage in Diabetes Mellitus and Atherosclerosis
The correlation between diabetes mellitus and ischemic stroke has been found by several authors and is a common experience in clinical practice. In agreement with previous studies, Matz et al. discovered a high prevalence of glucose metabolism disorders in patients with ischemic stroke, including new-onset diabetes mellitus. While diabetes is a known risk factor for the occurrence of ischemic stroke, it remains to be understood whether asymptomatic hyperglycemia has a potential role in causing a cerebrovascular event [
41,
42].
The frequency of T2D is three times higher in patients with ischemic stroke than in controls [
43].
Indeed, the risk of stroke increases from 150% to 400% in diabetics, and the extent of glycemic dyscontrol correlates directly with the risk of acute cerebrovascular accidents [
41,
44].
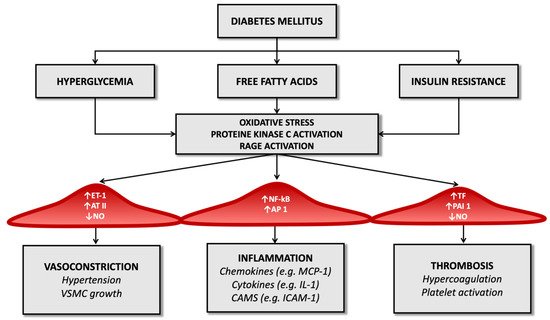
The altered metabolic framework that characterizes diabetes mellitus influences vascular changes. The most characteristic abnormalities certainly include chronic hyperglycemia, insulin resistance, and dyslipidemia: these factors can promote the atherosclerotic structure and induce cellular dysfunction at various levels (like endothelial dysfunction, smooth muscle cell alterations, platelet abnormalities, and coagulation alterations).
Concerning endothelial dysfunction, it should be remembered that endothelial cells represent the inner lining of the vascular lumen and perform, in addition to the mechanical function of coating, an endocrine role through the production of biologically active substances that regulate various functions: nitric oxide, prostaglandins, endothelin-1, angiotensin II, and other reactive oxygen species. Nitric oxide dilates the vessels, inhibits platelet activation, reduces the proliferation of smooth muscle cells, and reduces the process of diapedesis of leukocytes. The combination of these phenomena aims to limit the atherosclerotic pattern, ensuring the vascular system’s integrity [
45,
46,
47].
Several studies have shown that T2D alters endothelial function through a cascade of molecular events that precede atherosclerotic plaque formation but promote its appearance. Constant hyperglycemia inhibits the enzyme eNOS (endothelial nitric oxide synthase) and thus cause the reduction of nitric oxide and the stimulation of the production of reactive oxygen species, including superoxide anion (O
2−) [
48]; superoxide anion neutralizes nitric oxide by producing the toxic ion peroxynitrite, which uncouples the eNOS enzyme by oxidizing its cofactor, tetrahydrobiopterin [
49].
Another mechanism that limits the production of nitric oxide is related to insulin resistance: the reduced action of insulin on adipocytes leads to an increased release of free fatty acids from adipose tissue [
50] with consequent activation of the pathway of protein kinase C, then inhibition of phosphatidylinositol-3 (PI-3) kinase and increase of reactive oxygen species at the expense of nitric oxide [
51].
While, from what has been said above, T2D reduces the production of mediators of vasodilation, it is also true that in diabetic patients, the production of vasoconstrictive substances increases, including, crucially, endothelin-1.
Endothelin-1 has a dual effect: it promotes vasoconstriction through its action on smooth muscle and activates renal salt and water retention, resulting in activation of the renin-angiotensin-aldosterone system and thus smooth muscle hypertrophy [
52].
The concentration of endothelin-1, among other things, increases in response to the insulin-mediated effects of increased gene expression and receptor synthesis and as a consequence of the increased presence of glycosylation products, so its vaso-active effects in diabetes are multiple and complex [
53].
Moreover, endothelial cells regulate the cell transit through the vessel wall producing chemotactic adhesion molecules: monocytes, once reaching the subendothelial space, phagocytize oxidized LDL and become foamy cells, the initial substrate of atherosclerotic lesions [
23].
Advanced glycosylation end-products (AGEs) also amplify endothelial cell damage: high serum glucose levels produce a process of glycation and glycosylation of proteins at the extracellular level, leading to their accumulation. These molecular products cause the increased expression of adhesion molecules on endothelial cells, promote the migration of monocytes/macrophages towards the forming atherosclerotic plaque and promote the release of inflammatory cytokines by histiocytes. They also contribute to changes in the extracellular matrix and are a risk factor for plaque rupture [
54,
55] (
Figure 1).
Figure 1. Diabetes’ mechanisms of vascular damage.
In the diabetic patient, therefore, all these molecular pathways (hyperglycemia, increased oxidative stress, and activation of receptors for advanced products of glycosylation) promote the expression of the gene for the nuclear factor kappa-light-chain-enhancer of activated B cells (Nf-kB): this leads to increased production of inflammatory mediators (such as interleukin-1) and adhesion molecules for leukocytes that favor atherogenesis [
17,
56,
57].
Thus, endothelial dysfunction represents a significant factor in the pathogenesis of ischemic stroke and is a shared effect of diabetes mellitus and atherosclerosis, the ‘accelerator pedal’ on which both these diseases press.
The reduced bioavailability of nitric oxide and the prevalence of molecules mediating vasoconstriction in people with diabetes results in altered smooth muscle cell function: infusion of angiotensin II or endothelin-1, in fact, in these patients, results in a lower vasoconstriction effect than in healthy controls [
58,
59,
60].
Among other things, diabetic patients have autonomic dysfunction that modifies peripheral vascular resistance through mechanisms that are not yet fully known and this, together with the above, constitutes an essential element of vascular damage [
61].
The altered metabolic pattern generated by diabetes also results in structural and functional alterations in smooth muscle cells. In non-diabetic subjects, during the atherogenic process, muscle cells migrate from the intermediate layer of the vascular wall to the forming atherosclerotic plaque, replicating and contributing to the extracellular matrix’s constitution. In people with T2D, an increased migration capacity of smooth muscle cells has been demonstrated in vitro. Still, these cells are reduced in complicated atherosclerotic plaques [
62] since hyperglycemia determines modifications on the oxidation process of low-density lipoprotein (LDL) that favor their apoptosis [
63].
The progression of atherosclerosis and the risk of plaque rupture in diabetic patients is also linked to platelet dysfunction generated by the disease: the concentration of glucose inside platelets, like endothelial cells, does not depend on the action of insulin but the extracellular concentration of glucose. The increased intracellular availability of glucose causes activation of protein kinase C, reduced nitric oxide production, and increased reactive oxygen species, as mentioned above about endothelial cells [
64].
The result is impaired platelet function, altered homeostasis of intracellular calcium, and dysregulation of the thromboxane synthesis process. In addition, in diabetic patients, increased surface expression of glycoprotein Ib (which mediates interaction with von Willebrand factor) and glycoprotein IIb/IIIa, which regulates interaction with fibrin and leads to a significant increase in potential thrombotic risk [
65].
Platelet alteration is only one aspect of the alterations inherent in the coagulation process. For example, several studies have observed that in diabetic patients, there is reduced fibrinolytic activity due to the high concentration of the tissue plasminogen activator inhibitor type 1 both in atheromatous plaques and in arteries not affected by the atheromatous process [
66].
Diabetes also determines an imbalance between factors that regulate the hemostatic balance, favoring the increased availability of tissue factor and factor VII of the coagulation cascade (which have a potent procoagulant activity) and determining a reduction in serum levels of anticoagulant factors such as protein C and antithrombin III (
Figure 1). It seems that these alterations depend directly on the condition of hyperglycemia that characterizes T2D and partly on the cleavage products of proinsulin in the process of transformation to insulin [
67].
Chronic inflammation is another determinant of thrombotic risk and is a common feature of atherosclerosis and diabetes mellitus. Increased inflammasome activity and high levels of nucleotide-binding oligomerization domain-like receptor 3 (NLRP3) have been documented in diabetic patients, together with increased serum levels of pro-inflammatory cytokines such as interleukin-1 beta and interleukin-18. One of the common aspects of diabetes and atherosclerosis in the pattern of inflammation is neutrophil extracellular trap activation, or NETosis, a particular type of cell death in macrophages through which chromatin is released into the extracellular space to trap and kill bacteria. This mechanism is typical of chronic inflammatory conditions and infections, and it has been observed that it can be promoted by hyperglycemia [
68].
Additionally, in animal models, NETosis has been shown to promote atherosclerosis so that, through anti-diabetic therapy, it is possible to limit the atherosclerotic burden by defining the process of atherosclerotic plaque genesis.
In addition to the already known mechanisms by which T2D promotes inflammation, recently, the high-mobility group box 1 protein (HMGB1), non-histone proteins that act as an alarm for the immune system to initiate tissue repair and host defence processes, have been increasingly studied. It appears that during the acute phase of stroke, these proteins migrate towards the extracellular space and are bound by Toll-like receptors (TLR-2, TLR-4) and receptors for advanced glycosylation end-products (RAGE) that activate the transcription factor Nf-kB and the synthesis of inflammatory cytokines that worsen outcome and prognosis [
69].
According to [
60], in diabetic patients, the excess presence of advanced glycosylation end-products (AGEs) and their receptors (RAGEs) is an unfavorable element, so the possibility of limiting the expression of HMGB1, especially in diabetics, is being investigated to limit the inflammatory pattern of ischemic stroke and to improve the prognosis of these patients.
Altogether, diabetes mellitus is a true generator of thrombotic risk and, therefore, a promoter of ischemic stroke: on the one hand, it activates molecular damage mechanisms leading to endothelial dysfunction and the progression of the atherosclerotic process, and on the other, it increases the thrombogenic risk through the induction of platelet dysfunction and the dysregulation of the coagulation cascade.
3. Epidemiology of Ischemic Stroke in T2DM Patients
Type 2 diabetes mellitus (T2DM) is one of the most common chronic pathologies, and in 2015 nearly 400 million subjects in the world were considered diabetic; the prevalence of this disease is supposed to increase up to 640 million people in the next 20 years [
70].
Stroke affects more than 500,000 persons every year in the United States and represents one of the most significant reasons for decease in the Western part of the world [
71]; in addition, ischemic stroke is the second most frequent complication of T2DM after coronary artery disease (CAD) [
72], and the second source of death after cancer in diabetic subjects [
73].
The Framingham Study showed that the incidence of ischemic stroke in diabetic patients was 2.5–3.5 times increased compared to the control group [
74]. Similar results have been observed in multiple research [
75,
76].
While cardioembolic stroke is more common in non-diabetic subjects, diabetes is correlated to cerebral ischemia caused by atherosclerosis [
77]; the endothelial dysfunction can easily explain this due to the persistent hyperglycemia and by the concomitant presence of other risk factors for atherosclerosis, such as hypertension and hyperlipidemia.
In an observational study, Mulnier et al. reported a rate of stroke of 11.9 per 1000 persons per year in subjects affected by DM, in the healthy group, this rate was 5.5 per 1000 person-year; the highest hazard ratio correlated with ischemic stroke was noticed in the 35–54 year group, in addition, the risk was significantly higher in women than in men [
78]. As observed by Selvin et al., there is a correlation between the levels of glycated hemoglobin (HbA1c) and the chance of cerebral ischemia [
79]. Furthermore, proteinuria (>300 mg/dL) has been associated with an increased risk of ischemic stroke, even if it does not seem to influence the prognosis [
80].
T2DM is rarely an isolated condition; in the majority of the cases, it is associated with other well-known risk factors for ischemic stroke, such as dyslipidemia and hypertension. Consequently, it is necessary to operate on these pathologies to reduce the chance of cerebral ischemia in diabetic subjects.
Kearney et al. performed a meta-analysis of 14 randomized trials including more than 18,000 subjects affected by diabetes mellitus; statin therapy led to a significant reduction of the risk of ischemic stroke; this reduction was more marked in diabetic patients than in the control group [
81].
As far as it concerns hypertension, the UK Prospective Diabetes Study (UKPDS) showed that the reduction of 10 mmHg in systolic blood pressure resulted in a more than 40% decrease in stroke incidence [
82]. Furthermore, treatment with indapamide plus perindopril was associated with a 38% diminution of the stroke risk in patients affected by diabetes mellitus, as reported by the Perindopril Protection Against Recurrent Stroke Study (PROGRESS) [
83].
Moreover, Lichtman et al. reported diabetes as an independent risk factor for cerebral ischemia in the first six months after acute coronary syndrome [
84]; the same finding was observed after coronary artery bypass grafting [
85].
Diabetes mellitus is a common comorbidity in subjects affected by an ischemic stroke. Up to 20% of stroke patients have diabetes [
86,
87,
88]; furthermore, Gray et al. reported a new diagnosis of diabetes by 12 weeks after the stroke in nearly 20% of the subjects [
89].
Multiple studies reported the significant association between DM and lacunar stroke [
90]. This finding is quite plausible because hyperglycemia, together with hypertension, plays a crucial role in the process of lipohyalinosis, which is the leading cause of small vessel disease. Nevertheless, some studies did not find this association [
91,
92].