Staphylococcus aureus is a fatal Gram-positive pathogen threatening numerous cases of hospital-admitted patients worldwide. The emerging resistance of the pathogen to several antimicrobial agents has pressurized research to propose new strategies for combating antimicrobial resistance. Novel strategies include targeting the virulence factors of S. aureus. One of the most prominent virulence factors of S. aureus is its eponymous antioxidant pigment staphyloxanthin (STX), which is an auspicious target for anti-virulence therapy.
- anti-virulence
- staphyloxanthin
- Staphylococcus aureus
- CrtM
- CrtN
- MRSA
1. Naturally Occurring STX Inhibitors
1.1. Flavonoids
1.2. Rhodomyrtone
1.3. Marine Bioresource: Chitosan
1.4. Pogostemon heyneanus and Cinnamomum tamala Essential Oils
1.5. 2-Hydroxy-4-Methoxybenzaldehyde (HMB)
1.6. Myrtenol
1.7. Euphorbia tirucalli Latex
1.8. Schinus terebinthifolia Leaf Lectin
1.9. Callistemon citrinus Skeels
1.10. The Essential Oil of Eugenia brejoensis L. (Myrtaceae)
1.11. Ginkgo biloba Exocarp Extract
1.12. Carvacrol
1.13. Thymol
1.14. Hesperidin
2. Chemically Synthesized Inhibitors
2.1. Indole and Halogenated Indoles
2.2. Tetrangomycin Derivatives
2.3. Repurposing FDA-Approved Drugs
2.3.1. Cholesterol-Lowering Agents
In accordance with Oldfield, the catalysis of two farnesyl diphosphate (FPP) molecules into presqualene diphosphate by CrtM is considered the first basic step in the biosynthesis of staphyloxanthin by S. aureus. The resemblance in structure between CrtM and human squalene synthase (SQS), responsible for cholesterol biosynthesis in humans, aided the repurposing of some cholesterol-lowering agents into STX blockers (Figure 1). The utilization of cholesterol inhibitors as anti-virulence drugs has caused a significant inhibition to S. aureus virulence through directly inhibiting the carotenoid pigment production, rendering the treated strains vulnerable to the oxidative stress of human innate immunity and hence rapid clearance of the microorganism [57].
In 2008, a study conducted by Liu et al. suggested evident structural resemblance between S. aureus CrtM and human SQS. In attribution to the deduction, the analogy between the biosynthetic pathway of cholesterol in humans and that of STX production in S. aureus could be further studied. One cholesterol-lowering agent has been previously tested as a successful STX inhibitor that rendered S. aureus susceptible to oxidative stress of the human neutrophils in a mouse model after depigmenting the strain [16]. Later in 2009, Song et al. evaluated the possibility of inhibiting CrtM by potent phosphonosulfonates especially with halogen substitution and were able to prove their inhibitory effect on STX production with no effect against human squalene synthase [58].
Similarly, phosphonoacetamides were tested for STX inhibition in vitro and in a mouse infection model where a significant inhibition of disease progression was evident in the latter. X-ray crystallography revealed the most active compound to be N-3-(3-phenoxyphenyl) propylphosphonoacetamide [59].
With focus on cholesterol-lowering drugs, lapaquistat acetate and squalestatins are reported to inhibit SQS in humans and hence their cholesterol-lowering activity. Molecular docking analysis was performed to detect the mode of binding of lapaquistat acetate and squalestatin analogs to CrtM enzyme of S. aureus (Figure 6). Molecular docking confirmed the involvement of specific target sites on the CrtM enzyme when introduced to the respective SQS inhibitors. Among the most prominent target residues were His18, Arg45, Asp48, Asp52, Tyr129, Gln165, Asn168 and Asp172 [60].
Figure 6. Molecular docking analysis showing 2D (on the right panel) and 3D (on the left panel) representation of interaction patterns of lapaquistat acetate with dehydrosqualene synthase receptor [25].
2.3.2. Glyceryl Trinitrate (GTN)
GTN is a well-known medication for the treatment of cardiovascular diseases. Not only does GTN provide renowned anti-angina effects, but in a recent study it was also evident that this drug is capable of STX inhibition, biofilm disruption and oxidative stress resistance in S. aureus strains. Regarding in silico studies, it is reported that GTN binds with high affinity to CrtM which explains its marked STX inhibition activity. Thus, GTN could be a promising antipathogenic candidate against S. aureus [61].
2.3.3. Diclofenac
Abbas et al. proposed that the renowned anti-inflammatory drug diclofenac possesses a notable anti-virulence effect against MRSA strains. The assumption was based on the discovery of the drug antipathogenic activity against Pseudomonas aeruginosa and Proteus mirabilis. In this work, diclofenac exerted an anti-STX production activity against MRSA clinical isolates at sub-MICs that reached 8–57.2% when compared to controls. Diclofenac treatment resulted also in decreased biofilm formation (22.67–70%) and noteworthy inhibition of hemolysin activity (5.4–66.34%). The phenotypic results were further confirmed by transcriptomic analysis using quantitative real time PCR that revealed marked downregulation of the previously tested virulence genes. Hence, diclofenac therapy along with other antimicrobials is recommended as an anti-virulence treatment against deleterious MRSA strains [62].
2.3.4. Domperidone
Domperidone, an FDA-approved antiemetic drug, was studied by El-Ganiny et al. to detect its potential anti-virulence activity against S. aureus. Significant inhibition of the carotenoid pigment of S. aureus was detected using sub-inhibitory concentrations of domperidone to reach 76.4–81.23% at 1/8 MIC (9.8 μg/mL) and 1/4 MIC (19.5 μg/mL), respectively. Furthermore, the inhibition of the biofilm formation using sub-inhibitory concentrations of domperidone reached 84.37% at 1/4 MIC and 80.16% at 1/8 MIC. Gene expression analysis using qRT-PCR further confirmed the phenotypic results revealing decreased expression levels of virulence genes such as CrtM, SigB, SarA, AgrA, hla, fnbA, and icaA by domperidone treatment [63].
2.3.5. Candesartan
Candesartan, a widely used drug in the treatment of high blood pressure, is now being re-studied for inherent anti-virulence characteristics against S. aureus. Candesartan’s ability to inhibit the antioxidant carotenoid pigment of S. aureus was evaluated using sub-inhibitory concentrations of the drug to yield pigment inhibition of 85.57% at 1/4 MIC (1.2 μg/mL) and 80.57% at 1/8 MIC (0.6 μg/mL), respectively. Furthermore, the inhibition of the biofilm formation using sub-inhibitory concentrations of domperidone reached 87.63% at 1/4 MIC and 71.5% at 1/8 MIC. Quantitative gene analysis revealed downregulation of virulence genes of S. aureus with the greatest inhibition activity against CrtM, sigB, sarA, agrA, hla and icaA genes [63].
2.3.6. Antifungal Agents
Feifei et al. reported that the antifungal naftifine exerted a potent STX inhibitory activity via competitive inhibition of CrtN enzyme when tested on MSSA cells. The drug was capable of inhibiting the carotenoid pigment without affecting the growth of MSSA cells in a dose-dependent manner (up to 0.2 mM ~64.8 μg/mL) [64]. Later in 2020, Jing et al. proposed the synergistic role of naftifine to photodynamic antimicrobial chemotherapy (PACT) against S. aureus. The aiding role of naftifine is believed to be due to its inhibitory activity to STX that scavenges the reactive oxygen species (ROS) generated by PACT. Hence, the notorious antifungal resulted in an enhanced PACT activity when incubated with S. aureus cells at a concentration of 10 μM [65].
A recent study carried by El-Ganiny et al. focused on miconazole, which was reported to exhibit anti-virulence effects when studied against S. aureus standard strain (well-characterized strain with defined susceptibility or resistance profiles to the antimicrobial agents tested). In that study, 1/4 MIC (18.75 μg/mL) and 1/8 MIC (9.4 μg/mL) of miconazole were used to inhibit multiple virulence characteristics of S. aureus. STX inhibition reached 76.43–83.93% upon treatment with the indicated sub-inhibitory concentrations of the drug. Furthermore, the inhibition of the biofilm formation using sub-inhibitory concentrations of domperidone reached 90% at 1/4 MIC and 86.84% at 1/8 MIC. Transcriptomic analysis using qT-PCR divulged the reduced expression of CrtM, SigB, SarA, AgrA, hla, FnbA, and IcaA [63].
2.4. Newly Discovered CrtN Inhibitors
2.4.1. 5 m Analog
Wang et al. have previously revealed that CrtN is a promising target for anti-virulence therapy. They have further disclosed the ability of the famous antifungal naftifine to drastically abolish the carotenoid pigment production in S. aureus species. The discovery of 5 m, a novel type of Benzofuran-derived CrtN inhibitor, has recently followed in the footsteps of the repurposing strategy of naftifine. As a result, the analogy has reflected a typical effect of the 5 m analog on S. aureus Newman and three other methicillin-resistant strains with low IC50 values ranging from 0.38–5.45 nM. The treated cells were rendered susceptible to immune clearance and their virulence was markedly weakened [66].
2.4.2. Compound NP16
A newly discovered compound termed NP16 has showed a potent activity as a CrtN inhibitor in S. aureus strains. Consequently, notable interruption to the golden carotenoid pigment biosynthesis was evident that further caused an increased vulnerability to oxidative stress and neutrophil killing in vivo [67].
2.4.3. 1,4-Benzodioxan-derivatives
In 2018, 38 1,4-benzodioxan-derived CrtN inhibitors were synthesized to combat the downsides of the leading compound 4a. Derivative 47 exhibited a remarkable CrtN inhibitory effect with higher potency than 4a (pigment inhibition in S. aureus Newman: IC50 = 270.4 ± 43.8 nM by compound 47 vs. IC50 = 1.9 nM by compound 4a) in addition to enhanced water solubility. The sensitization effect of derivative 47 on MRSA strains was reported to be quite significant and successfully facilitated immune clearance in vitro [68].
2.5. Others
Farnesol
Candida albicans and S. aureus are among the most commonly known opportunistic pathogens that usually co-exist in mixed biofilms [69]. Both pathogens are often isolated together from hospital-admitted patients [70]. In a recent study, the role of C. albicans-secreted quorum sensing (QS) molecule (farnesol) was assessed against S. aureus cells. The study mimicked a mixed biofilm of C. albicans with S. aureus cells by repetitive exposure of S. aureus to farnesol. The sensitized S. aureus cells revealed significant inhibition of STX. The findings of transcriptional analysis further displayed marked changes in the expression of global regulators involved in resistance to oxidative stress. Unfortunately, the activation of stress-response mechanisms in S. aureus boosted its tolerance to intracellular killing and ROS. The pigment inhibition effect was reasoned then by proposing a theoretical binding model that indicated the binding of farnesol to CrtM enzyme causing blockage of the biosynthetic pathway of STX due to its structural resemblance to the substrate of CrtM. Those findings illustrate the role of the fungal-secreted QS mediator that could successfully elicit oxidative stress on S. aureus through thiol-based redox system activation. Moreover, the results of the previous study reported that depigmentation mediated by STX inhibitors was considered a transient conditional state, as upon the gradual removal of farnesol, gradual recovery of the pigment was observed in comparison with the control cells [71].
Table 1. Chemical structures of various staphyloxanthin inhibitors.
|
STX Inhibitors |
Chemical Structure |
|
1. Flavone |
|
|
2. Myricetin |
|
|
3. Rhodomyrtone |
|
|
4. Chitosan |
|
|
5. E-nerolidol (derived from P. heyneanus and C. tamala essential oils) |
|
|
6. 2-hydroxy-4-methoxybenzaldehyde (HMB) [30] |
|
|
7. Myrtenol |
|
|
8. Euphol (derived from Euphorbia tirucalli latex) [72] |
|
|
9. cis-β-terpineol (derived from Schinus terebinthifolia leaf lectin) [37] |
|
|
10. Pulverulentone A (C1) (derived from Callistemon citrinus Skeels) [40] |
|
|
11. δ-cadinene (derived from the essential oil of Eugenia brejoensis L.) [41] |
|
|
12. Ginkgoic acid (derived from Ginkgo biloba exocarp extract) [73] |
|
|
13. Carvacrol |
|
|
14. Thymol |
|
|
15. Hesperidin |
|
|
16. 7-benzyloxyindole |
|
|
17. Tetrangomycin |
|
|
18. 2-isopropyl-naphtho[2,3-b] furan-4,9-dione (tetrangomycin derivative) |
|
|
19. Lapaquistat acetate |
|
|
20. Squalestatin |
|
|
21. Glyceryl trinitrate |
|
|
22. Diclofenac sodium |
|
|
23. Domperidone |
|
|
24. Candesartan |
|
|
25. Naftifine |
|
|
26. Miconazole |
|
|
27. 5M analog [66] |
|
|
28. NP16 [67] |
|
|
29. Derivative 47 [68] 4-Benzodioxine-7-methyl)-N-methyl-5-(4-trifluorophenyl)prop-2,4-dien-1-amine Hydrochloride |
|
|
30. Farnesol |
|
Chemical structures are from Reaxys website via Egyptian Knowledge Bank (EKB) 2022 (https://081291uul-1104-y-https-www-reaxys-com.mplbci.ekb.eg/).(accessed 14th February, 2022)
Table 2. Structure activity relationship (SAR) of previously studied STX inhibitors.
|
Compound |
Structure Activity Relationship (SAR) |
|
Flavone |
Carbonyl moiety is crucial for activity, interacts with adjacent amino acid residues in CrtM receptor by conventional hydrogen bond. Yet, the exact mechanism of action for anti-virulence activity remains to be determined. [10,20,74] |
|
Myricetin |
Hydroxyl moiety enhances the binding affinity to adjacent amino acid residues of the CrtM receptor through conventional hydrogen bonds. Carbonyl group is essential for activity, binds to adjacent amino acids of the receptor via hydrogen bonds[21,74]. |
|
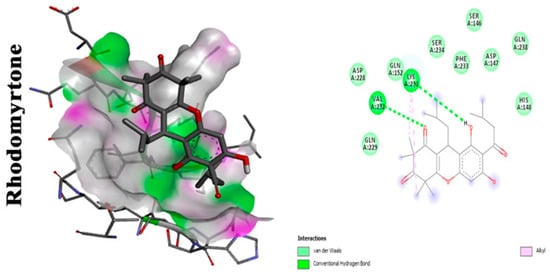
Rhodomyrtone |
Alkyl interactions with CrtM receptor at CH3 terminals. Carbonyl moiety is crucial for activity, interacts with VAL in CrtM receptor by hydrogen bond. Hydroxyl moiety enhances the binding affinity to LYS residues through conventional hydrogen bonds [24,25]. |
|
E-nerolidol |
The alcoholic moiety is essential for activity, binds to CrtM through hydrogen bond. The backbone of the structure interacts with the hydrophobic pocket of the receptor [28]. |
|
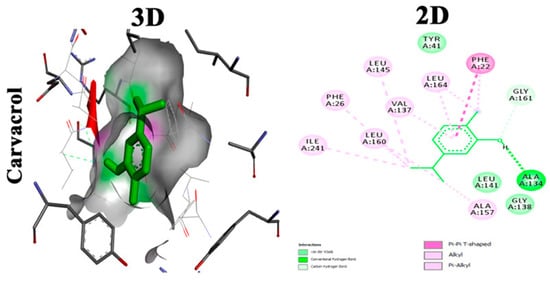
Carvacrol |
Essential phenolic hydroxyl group for activity, binds with conventional hydrogen bond to CrtM. Oxygen involved in the hydroxyl group interacts with GLY A:161 through carbon hydrogen bond. Phenyl ring binds by Pi–Pi T-shaped bond to PHE A:22. Terminal methyl groups bind to ALA A:157, ILE A:241 and PHE A:22 [25]. |
|
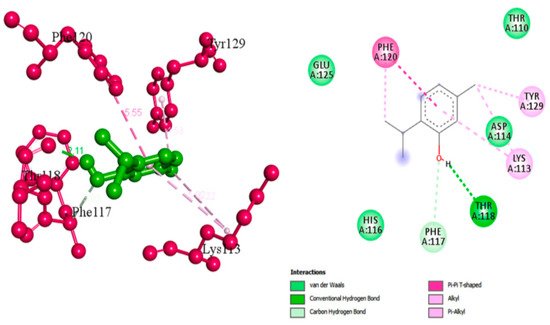
Thymol |
Essential phenolic hydroxyl group for activity, binds with conventional hydrogen bond to CrtM. Oxygen involved in the hydroxyl group interacts with PHE A:117 through carbon hydrogen bond. Methyl terminals bind to TYR A:129, LYS A:113 and PHE A:120 through alkyl interactions [49]. |
|
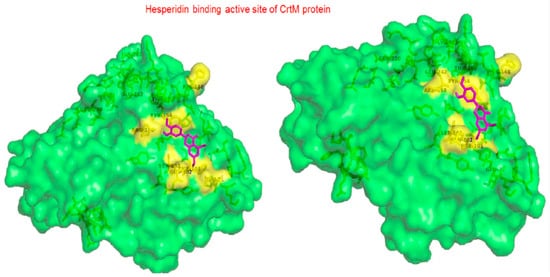
Hesperidin |
In case of CrtM, hesperidin actively interacts through (Arg 158, Tyr 154 and Gln 102). Carbonyl moiety interacts with amino acid residues via hydrogen bond. Hydroxyl groups enhances the activity [51]. |
|
Tetrangomycin |
Hydrogen acceptor groups are crucial for activity. Lipophilic moieties decorating the naphthoquinone ring enhance STX inhibition [55,56]. |
|
7-benzyloxyindole |
The presence of ether group (acidic moiety) enhances the activity of the compound. The addition of a second hydrophobic ring enhances the activity [53,75]. |
|
2-isopropylnaphtho [2,3-b]furan-4,9-dione |
Hydrogen acceptor groups are crucial for activity. Lipophilic moieties decorating the naphthoquinone ring enhance STX inhibition [55,56]. |
|
Lapaquistat acetate |
Carbonyl groups interact with adjacent amino acid residues in CrtM receptor via conventional hydrogen bonds. Aromatic ring interaction with adjacent amino acid residues via alkyl interactions. Methyl group interacts with PHE A:267 on the receptor via Pi–alkyl interaction [25]. |
|
Squalestatin |
Interaction of carbonyl groups, hydroxyl groups and aromatic benzene ring with His18, Arg45, Asp48, Asp52, Tyr129, Gln165, Asn168 and Asp172 residues on CrtM receptor [60]. |
|
Glyceryl trinitrate |
GTN has nine hydrogen bonds with Arg45, Tyr129, Gln165, Asn168, Val 133 and Tyr248 electrostatic interaction with Arg45 and Asp48 and pi-cation interaction of the nitrogen atom with Tyr183 [61]. |
|
Naftifine |
The naphthalenyl moiety of NTF is not indispensable for pigment inhibitory activity, the N-methyl group is critical for high potency on CrtN receptor [66]. |
|
5M analog |
The naphthalenyl moiety of NTF is not indispensable for pigment inhibitory activity, the N-methyl group is critical for high potency, the 4-substituted phenyl moiety is critical for high potency on CrtN receptor. The para-position is the best substituted position at the phenyl ring [66]. |
|
Derivative 47 |
The N-methyl group is critical for high potency. Unsubstituted alkenyl linker was critical for improving pigment inhibitory activity. The 4-substituted phenyl moiety is critical for high potency on CrtN receptor. The para-position is the best substituted position at the phenyl ring [66,68]. |
-
Naturally Existing Susceptible Variant
In 2013, the hypothesis of linking virulence attenuation to pigment inhibition was further investigated. Clonal complex 75 of S. aureus (S. argenteus) revealed a lack of STX regulating operon, CrtOPQMN, which divulged its inability to produce the carotenoid pigment and hence its vulnerability to ROS and neutrophil clearance when experimented on in vitro as well as in vivo. On the other hand, deliberately transformed cells with pTX crtOPQMN lead to increased resistance to oxidative stress [76].
-
Reversing STX Inhibition
4.1. Inter-Species Communication
Quorum sensing (QS) or cell–cell communication is a pivotal process governed by some secreted mediators that allow the recognition of one species to the other and behavioral coordination in accordance. Hence, the secreted molecules can remarkably alter the cell physiology of the species present in a polymicrobial domain [77]. S. aureus strains are naturally present as yellow and white colony variants. It is believed that bacterial pigments are usually synthesized in stressful conditions.
Owing to the fact that quorum sensing can control pigment production in a variety of bacterial cells, as discussed earlier, coinfection with C. albicans resulted in inhibition of S. aureus STX pigment due to the secretion of farnesol. However, co-infection with Pseudomonas aeruginosa resulted in increased production of STX and catalase in the white variants of S. aureus through QS. The induced carotenoid pigment by P. aeruginosa conferred polymyxin resistance and hydrogen peroxide endurance upon the respective S. aureus cells. Thus, it is assumed that the STX biosynthesis mechanism is always present in S. aureus variants which can be induced by the chemical signaling molecules of P. aeruginosa and inter-species interaction [78].
4.2. Inactivation of Catabolite Control Protein E (CcpE)
Metabolism in S. aureus is crucially regulated by the catabolite control protein E (CcpE) where catalase binds to stimulate the expression of almost 4.7% of the S. aureus genetic material. Surprisingly, it was found that the inactivation of CcpE has led to enhancing S. aureus virulence to include increased STX production, resistance to whole-blood killing and iron scavenging. CcpE is now known as S. aureus metabolic sensor that allows the strains to regulate virulence expression [79].
-
Genetic Manipulation
5.1. msaABCR Operon
- aureus has developed an arsenal of defense mechanisms mediated by a complex grid of regulators that allows the strains to survive under oxidative stress. According to Pandey et al., msaABCR operon was depicted to be a stress regulator in S. aureus cells which conveyed the character of resistance to antibiotics and oxidative stress and aided the generation of dormant cells of S. aureus that in turn exhibited multidrug tolerance. Transcriptional analysis revealed downregulation of multiple genes responsible for resistance against oxidative stress in response to the deletion of msaABCR operon. Genes involved in STX as well as the ohr gene (responsible for the cellular defence against organic hydroperoxides) were also downregulated upon the deletion of the operon. Moreover, it has been reported that MsaB, a protein product of the msaABCR operon, can regulate the expression of crtOPQMN operon (responsible for the production of the golden pigment) and the ohr gene as well as its repressor, resulting in a controlled resistance to oxidative stress in vitro. This study casts a light on the crucial role of the msaABCR operon and its protein product MsaB in defending S. aureus against unfavorable environmental conditions as oxidative stress. Based on this discovery, the operon could be a prospective target for antipathogenic treatment against the recurrent staphylococcal infections [80].
5.2. SigB
The expression of the golden yellow pigment is governed by the global stress response regulator SigB. The presence of SigB allows S. aureus to increase the expression of multiple resistance genes including the ones responsible for oxidative stress resistance manifested in the eponymous STX pigment. In a recent study, radiation survival of S. aureus was examined to detect the protective role of SigB against radiation damage. Mutant strain cells of S. aureus lacking the carotenoid pigment (crt enzymes-deficient) were subjected to different types of radiation. The crt-mutant cells were reported to be threefold more susceptible to UV radiation than the wild-type strain [81].
-
Possibility of an Emerging Resistance to Anti-virulence Therapy
The target of anti-virulence agents is to disrupt the pathogen’s virulence without affecting its growth or viability, thus preserving the normal microbiota, avoiding selective pressure on the bacteria and consequently reducing the emergence of antimicrobial resistance. However, several in vitro studies observed that there could be an emerging resistance to certain anti-virulence agents, but not all agents have the same risk of resistance [82–84]. Anti-virulence agents targeting a specific mechanism of virulence may have a limited probability of resistance compared to other agents targeting regulatory genes that may greatly affect multiple virulence factors. The emergence of resistance could be attributed to selective pressure on microbial subpopulation; however it is almost very weak in comparison with conventional antibiotics [85].
-
Conclusions
In this review we have discussed several naturally occurring as well as chemically synthesized STX inhibitors, hence avoiding selective pressure on S. aureus strains as a new approach to combat bacterial infections without drug resistance development. The significance of QS in regards to upregulation and downregulation of STX biosynthesis has been confirmed. Anti-virulence therapy is a novel approach towards a bacterial resistance-free regimen for a variety of infections. This approach is currently under preclinical investigations and it is recommended to proceed to clinical trials on human models after succeeding with in vitro and in vivo mouse models as well as invertebrate models. The STX inhibitors are considered potential anti-S. aureus agents particularly for the control of MRSA, a life-threatening pathogen of clinically relevant importance.
Author Contributions: Conceptualization. R.A.E.-M. wrote the first draft of the manuscript. S.E.S., H.M.E., I.S.Y. and K.M.A. revised the manuscript and revised the literature. S.E.S., H.M.E. and K.M.A. supervised the whole study and revised the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding: This research was funded by the Research Center for Advanced Materials Science (RCAMS) at King Khalid University (KKU/RCAMS/G012-21) and for the Deputyship for Research and Innovation, Ministry of Education, in Saudi Arabia for funding this research work through the project number: (IFP-KKU-2020/10).
Acknowledgments: The authors express their appreciation to the Department of Microbiology, Faculty of Pharmacy, Misr International University (MIU), and Department of Microbiology and Immunology, Faculty of Pharmacy, Ain Shams University (ASU), Cairo, Egypt, and to the Research Center for Advanced Materials Science (RCAMS) at King Khalid University (KKU/RCAMS/G012-21). Additionally, the authors extend their appreciation to the Deputyship for Research and Innovation, Ministry of Education, in Saudi Arabia for funding this research work through the project number: (IFP-KKU-2020/10).
Conflicts of Interest: The authors declare no conflict of interest.
References
- Vandendriessche, S.; Vanderhaeghen, W.; Soares, F.V.; Hallin, M.; Catry, B.; Hermans, K.; Butaye, P.; Haesebrouck, F.; Struelens, M.J.; Denis, O. Prevalence, risk factors and genetic diversity of methicillin-resistant Staphylococcus aureus carried by humans and animals across livestock production sectors. Antimicrob. Chemother. 2013, 68, 1510–1516.
- Federspiel, J.J.; Stearns, S.C.; Peppercorn, A.F.; Chu, V.H.; Fowler, V.G. Increasing US rates of endocarditis with Staphylococcus aureus: 1999–2008. Intern. Med. 2012, 172, 363–365.
- Sacco, K.A.; Termine, A.; Seyal, A.; Dudas, M.M.; Vessicchio, J.C.; Krishnan-Sarin, S.; Jatlow, P.I.; Wexler, B.E.; George, T.P. Effects of cigarette smoking on spatial working memory and attentional deficits in schizophrenia: Involvement of nicotinic receptor mechanisms. Gen. Psychiatry 2005, 62, 649–659.
- Kavanagh, K.T.J.A.R.; Control, I. Control of MSSA and MRSA in the United States: Protocols, policies, risk adjustment and excuses. Resist. Infect. Control 2019, 8, 1–8.
- Casadevall, A.; Pirofski, L.-A. Host-pathogen interactions: Redefining the basic concepts of virulence and pathogenicity. Immun. 1999, 67, 3703–3713.
- Clauditz, A.; Resch, A.; Wieland, K.-P.; Peschel, A.; Götz, F.J.I. Staphyloxanthin plays a role in the fitness of Staphylococcus aureus and its ability to cope with oxidative stress. Immun. 2006, 74, 4950–4953.
- Cho, H.S.; Lee, J.-H.; Cho, M.H.; Lee, J. Red wines and flavonoids diminish Staphylococcus aureus virulence with anti-biofilm and anti-hemolytic activities. Biofouling 2015, 31, 1–11.
- Cegelski, L.; Marshall, G.R.; Eldridge, G.R.; Hultgren, S.J. The biology and future prospects of antivirulence therapies. Rev. Microbiol. 2008, 6, 17–27.
- Muñoz-Cazares, N.; García-Contreras, R.; Pérez-López, M.; Castillo-Juárez, I. Phenolic compounds with anti-virulence properties” In Phenolic Compounds-Biological Activity, eds M. Soto-Hernández, M. P. Tenango, and R. García-Mateos (London: IntechOpen), 2017, 139–167. DOI: 10.5772/66367
- Xue, L.; Chen, Y.Y.; Yan, Z.; Lu, W.; Wan, D.; Zhu, H. Resistance, D. Staphyloxanthin: A potential target for antivirulence therapy. Drug Resist. 2019, 12, 2151.
- Lennette, E.H. Manual of Clinical Microbiology; ASM Press: Washington, DC, USA, 1985.
- Siems, W.; Sommerburg, O.; Schild, L.; Augustin, W.; Langhans, C.D.; Wiswedel, I. β‐Carotene cleavage products induce oxidative stress in vitro by impairing mitochondrial respiration. FASEB J. 2002, 16, 1289–1291.
- Fiedor, J.; Burda, K.J.N. Potential role of carotenoids as antioxidants in human health and disease. Nutrients 2014, 6, 466–488.
- Beavers, W.N.; Skaar, E.P.J.P. Neutrophil-generated oxidative stress and protein damage in Staphylococcus aureus. Dis. 2016, 74(6), 1-15: doi: 10.1093/femspd/ftw060
- Coker, M.S.; Forbes, L.V.; Plowman-Holmes, M.; Murdoch, D.R.; Winterbourn, C.C.; Kettle, A.J. Interactions of staphyloxanthin and enterobactin with myeloperoxidase and reactive chlorine species. Biochem. 2018, 646, 80–89.
- Liu, C.-I.; Liu, G.Y.; Song, Y.; Yin, F.; Hensler, M.E.; Jeng, W.-Y.; Nizet, V.; Wang, A.H.-J.; Oldfield, E.J.S. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus J. Sci. 2008, 319, 1391–1394.
- Pelz, A.; Wieland, K.-P.; Putzbach, K.; Hentschel, P.; Albert, K.; Götz, F.J.J.O.B.C. Structure and biosynthesis of staphyloxanthin from Staphylococcus aureus. Biol. Chem. 2005, 280, 32493–32498.
- Liu, G.Y.; Essex, A.; Buchanan, J.T.; Datta, V.; Hoffman, H.M.; Bastian, J.F.; Fierer, J.; Nizet, V. Staphylococcus aureus golden pigment impairs neutrophil killing and promotes virulence through its antioxidant activity. Med. 2005, 202, 209–215.
- Wieland, B.; Feil, C.; Gloria-Maercker, E.; Thumm, G.; Lechner, M.; Bravo, J.-M.; Poralla, K.; Götz, F. Genetic and biochemical analyses of the biosynthesis of the yellow carotenoid 4, 4′-diaponeurosporene of Staphylococcus aureus. Bacteriol. 1994, 176, 7719–7726.
- Lee, J.-H.; Park, J.-H.; Cho, M.H.; Lee, J.J.C.M. Flavone reduces the production of virulence factors, staphyloxanthin and α-hemolysin, in Staphylococcus aureus. Microbiol. 2012, 65, 726–732.
- Silva, L.N.; da Hora, G.; Soares, T.; Bojer, M.S.; Ingmer, H.; Macedo, A.J.; Trentin, D.S. Myricetin protects Galleria mellonella against Staphylococcus aureus infection and inhibits multiple virulence factors. Rep. 2017, 7, 1–16.
- Galati, G.; O’brien, P.J. Potential toxicity of flavonoids and other dietary phenolics: Significance for their chemopreventive and anticancer properties. Free Radic. Biol. Med. 2004, 37, 287–303.
- Siriyong, T.; Ontong, J.C.; Leejae, S.; Suwalak, S.; Coote, P.J.; Voravuthikunchai, S.P. In vivo safety assessment of rhodomyrtone, a potent compound, from Rhodomyrtus tomentosa leaf extract. Rep. 2020, 7, 919–924.
- Leejae, S.; Hasap, L.; Voravuthikunchai, S.P. Inhibition of staphyloxanthin biosynthesis in Staphylococcus aureus by rhodomyrtone, a novel antibiotic candidate. Med. Microbiol. 2013, 62, 421–428.
- Selvaraj, A.; Valliammai, A.; Muthuramalingam, P.; Priya, A.; Suba, M.; Ramesh, M.; Karutha Pandian, S.J.A.O. Carvacrol targets SarA and CrtM of methicillin-resistant Staphylococcus aureus to mitigate biofilm formation and staphyloxanthin synthesis: An in vitro and in vivo approach. ACS Omega 2020, 5, 31100–31114.
- Younes, I.; Rinaudo, M.J.M.D. Chitin and chitosan preparation from marine sources. Structure, properties and applications. Drugs 2015, 13, 1133–1174.
- Rubini, D.; Banu, S.F.; Hari, B.N.V.; Devi, D.R.; Gowrishankar, S.; Pandian, S.K.; Nithyanand, P.J.F.; Toxicology, C. Chitosan extracted from marine biowaste mitigates staphyloxanthin production and biofilms of Methicillin-resistant Staphylococcus aureus. Food Chem. Toxicol. 2018, 118, 733–744.
- Rubini, D.; Banu, S.F.; Nisha, P.; Murugan, R.; Thamotharan, S.; Percino, M.J.; Subramani, P.; Nithyanand, P.J.M.P. Essential oils from unexplored aromatic plants quench biofilm formation and virulence of Methicillin resistant Staphylococcus aureus. Pathog. 2018, 122, 162–173.
- Veer, V.; Singh, L.J.T.B. Field evaluation of repellency of a polyherbal essential oil against blackflies and its dermal toxicity using rat model. Biomed. 2012, 29, 391–397.
- Kannappan, A.; Srinivasan, R.; Nivetha, A.; Annapoorani, A.; Pandian, S.K.; Ravi, A.V. Anti-virulence potential of 2-hydroxy-4-methoxybenzaldehyde against methicillin-resistant Staphylococcus aureus and its clinical isolates. Microbiol. Biotechnol. 2019, 103, 6747–6758.
- Rathi, N.; Harwalkar, K.; Jayashree, V.; Sharma, A.; Rao, N.N. 2-hydroxy-4-methoxybenzaldehyde, an astounding food flavoring metabolite: A review. AJPCR 2017, 10, 105–110.
- Younes, M.; Aquilina, G.; Castle, L.; Engel, K.-H.; Fowler, P.; Fernandez, M.J.F.; Fürst, P.; Gürtler, R.; Gundert-Remy, U.; Husøy, T.; et al. Scientific opinion on flavouring group evaluation 414 (FGE. 414): 2‐hydroxy‐4‐methoxybenzaldehyde. EFSA 2021, 19, e06883.
- Bejeshk, M.; Fekri, M.S.; Najafipour, H.; Rostamzadeh, F.; Jafari, E.; Rajizadeh, M.; Masoumi-Ardakani, Y. Anti-inflammatory and anti-remodeling effects of myrtenol in the lungs of asthmatic rats: Histopathological and biochemical findings. Allergol. Immunopathol. 2019, 47, 185–193.
- Selvaraj, A.; Jayasree, T.; Valliammai, A.; Pandian, S. Myrtenol attenuates MRSA biofilm and virulence by suppressing sarA expression dynamism. Microbiol. 2019, 10, 2027.
- Colasso, A.H.M.; Barros, T.F.; Figueiredo, I.F.; Carvalho, A.R., Jr.; Fernandes, E.S.; Uchoa, M.R.B.; da Silva, L.C.N. The latex of Euphorbia tirucalli inhibits staphyloxanthin production and protects Tenebrio molitor larvae against Staphylococcus aureus Nat. Prod. Res. 2020, 34, 3536–3539.
- Mali, P.Y.; Panchal, S. Euphorbia tirucalli L.: Review on morphology, medicinal uses, phytochemistry and pharmacological activities. Asian Pac. J. Trop. Biomed. 2017, 7, 603–613.
- Lima, I.M.D.S.F.; Zagmignan, A.; Santos, D.M.; Maia, H.S.; Silva, L.D.S.; Cutrim, B.D.S.; Vieira, S.L.; Filho, C.M.B.; de Sousa, E.M.; Napoleão, T.H.; et al. Schinus terebinthifolia leaf lectin (SteLL) has anti-infective action and modulates the response of Staphylococcus aureus-infected macrophages. Rep. 2019, 9, 1–14.
- Ramos, D.D.B.M.; Araújo, M.T.D.M.F.; de Lima Araújo, T.C.; dos Santos Neto, O.G.; de Silva, M.G.; Silva, Y.A.; Torres, D.J.L.; de Siqueira Patriota, L.L.; de Melo, C.M.L.; de Lorena, V.M.B.J.J.O.E. Evaluation of antitumor activity and toxicity of Schinus terebinthifolia leaf extract and lectin (SteLL) in sarcoma 180-bearing mice. Ethnopharmacol. 2019, 233, 148–157.
- Sutar, N.; Sutar, R.; Kumar, M.J.I.R.J.P.; Sci. Callistemon citrinus (bottle brush) an important medicinal plant: A review of its traditional uses, phytoconstituents and pharmacological properties. Indian J. Pharm. Sci. 2014, 1, 68–77.
- Shehabeldine, A.M.; Ashour, R.M.; Okba, M.M.; Saber, F.R.J.J.O.E. Callistemon citrinus bioactive metabolites as new inhibitors of methicillin-resistant Staphylococcus aureus biofilm formation. Ethnopharmacol. 2020, 254, 112669.
- da Silva, A.G.; Alves, R.C.C.; Filho, C.M.B.; Bezerra-Silva, P.C.; Santos, L.M.M.D.; Foglio, M.A.; Navarro, D.M.D.A.F.; Silva, M.V.D.; Correia, M.T.D.S. Chemical composition and larvicidal activity of the essential oil from leaves of Eugenia brejoensis Mazine (Myrtaceae). Essent. Oil Bear. Plants 2015, 18, 1441–1447.
- Bezerra Filho, C.M.; da Silva, L.C.N.; da Silva, M.V.; Løbner-Olesen, A.; Struve, C.; Krogfelt, K.A.; Correia, M.T.D.S.; Vilela Oliva, M.L. Antimicrobial and Antivirulence Action of Eugenia brejoensis Essential Oil in vitro and in vivo Invertebrate Models. Microbiol. 2020, 11, 424. https://doi.org/10.3389/fmicb.2020.00424.
- Cao, C.; Su, Y.; Gao, Y.; Luo, C.; Yin, L.; Zhao, Y.; Chen, H.; Xu, A. Ginkgo biloba exocarp extract inhibits the metastasis of B16-F10 melanoma involving PI3K/akt/NF-κB/MMP-9 signaling pathway. Based Complementary Altern. Med. 2018, 2018, 1-11:4969028. doi: 10.1155/2018/4969028.
- Diamond, B.J.; Shiflett, S.C.; Feiwel, N.; Matheis, R.J.; Noskin, O.; Richards, J.A.; Schoenberger, N.E. Ginkgo biloba extract: Mechanisms and clinical indications. Phys. Med. Rehabil. 2000, 81, 668–678.
- Wang, B.; Wei, P.-W.; Wan, S.; Yao, Y.; Song, C.-R.; Song, P.-P.; Xu, G.-B.; Hu, Z.-Q.; Zeng, Z.; Wang, C.; et al. Ginkgo biloba exocarp extracts inhibit S. aureus and MRSA by disrupting biofilms and affecting gene expression. Ethnopharmacol. 2021, 271, 113895.
- Can Baser, K.J.C. Biological and pharmacological activities of carvacrol and carvacrol bearing essential oils. Pharm. Des. 2008, 14, 3106–3119.
- Ghorani, V.; Alavinezhad, A.; Rajabi, O.; Mohammadpour, A.H.; Boskabady, M.H. Safety and tolerability of carvacrol in healthy subjects: A phase I clinical study. Drug Chem. Toxicol. 2021, 44, 177–189.
- Nagoor Meeran, M.F.; Javed, H.; Al Taee, H.; Azimullah, S.; Ojha, S.K. Pharmacological properties and molecular mechanisms of thymol: Prospects for its therapeutic potential and pharmaceutical development. Pharmacol. 2017, 8, 380.
- Valliammai, A.; Selvaraj, A.; Muthuramalingam, P.; Priya, A.; Ramesh, M.; Pandian, S.K. Staphyloxanthin inhibitory potential of thymol impairs antioxidant fitness, enhances neutrophil mediated killing and alters membrane fluidity of methicillin resistant Staphylococcus aureus. Pharmacother. 2021, 141, 111933.
- Man, M.-Q.; Yang, B.; Elias, P.M. Benefits of hesperidin for cutaneous functions. Based Complementary Altern. Med. 2019, 2019, 1-19: 2676307. doi: 10.1155/2019/2676307
- Vijayakumar, K.; Muhilvannan, S.; Vignesh, M.A. Hesperidin inhibits biofilm formation, virulence and staphyloxanthin synthesis in methicillin resistant Staphylococcus aureus by targeting SarA and CrtM: An in vitro and in silico approach. World J. Microbiol. Biotechnol. 2022, 38, 1–12.
- Dhuguru, J.; Skouta, R.J.M. Role of indole scaffolds as pharmacophores in the development of anti-lung cancer agents. Molecules 2020, 25, 1615.
- Lee, J.-H.; Cho, H.S.; Kim, Y.; Kim, J.-A.; Banskota, S.; Cho, M.H.; Lee, J. Indole and 7-benzyloxyindole attenuate the virulence of Staphylococcus aureus. Microbiol. Biotechnol. 2013, 97, 4543–4552.
- Lee, J.-H.; Kim, Y.-G.; Gwon, G.; Wood, T.K.; Lee, J. Halogenated indoles eradicate bacterial persister cells and biofilms. AMB Express 2016, 6, 1–12.
- Özakin, S.; Davis, R.W.; Umile, T.P.; Pirinccioglu, N.; Kizil, M.; Celik, G.; Sen, A.; Minbiole, K.P.; İnce, E. The isolation of tetrangomycin from terrestrial Streptomyces sp. CAH29: Evaluation of antioxidant, anticancer, and anti-MRSA activity. Chem. Res. 2016, 25, 2872–2881.
- Ribeiro, L.; Fumagalli, F.; Mello, R.; Froes, T.; da Silva, M.; Gómez, S.V.; Barros, T.; Emery, F.; Castilho, M.J.M.P. Structure-activity relationships and mechanism of action of tetragomycin derivatives as inhibitors of Staphylococcus aureus staphyloxanthin biosynthesis. Pathog. 2020, 144, 104127.
- Oldfield, E. Targeting isoprenoid biosynthesis for drug discovery: Bench to bedside. Chem. Res 2010, 43, 1216–1226.
- Song, Y.; Lin, F.-Y.; Yin, F.; Hensler, M.; Poveda, C.A.R.; Mukkamala, D.; Cao, R.; Wang, H.; Morita, C.; Gonzalez-Pacanowska, D.; et al. Phosphonosulfonates are potent, selective inhibitors of dehydrosqualene synthase and staphyloxanthin biosynthesis in Staphylococcus aureus. Med. Chem. 2009, 52, 976–988.
- Song, Y.; Liu, C.-I.; Lin, F.-Y.; No, J.H.; Hensler, M.; Liu, Y.-L.; Jeng, W.-Y.; Low, J.; Liu, G.Y.; Nizet, V.; et al. Inhibition of staphyloxanthin virulence factor biosynthesis in Staphylococcus aureus: In vitro, in vivo, and crystallographic results. Med. Chem. 2009, 52, 3869–3880.
- Kahlon, A.K.; Roy, S.; Sharma, A. Dynamics. Molecular docking studies to map the binding site of squalene synthase inhibitors on dehydrosqualene synthase of Staphylococcus aureus. Biomol. Struct. Dyn. 2010, 28, 201–210.
- Abbas, H.A.; Elsherbini, A.M.; Shaldam, M. Glyceryl trinitrate blocks staphyloxanthin and biofilm formation in Staphylococcus aureus. Health Sci. 2019, 19, 1376–1384.
- Abbas, H.A.; Atallah, H.; El-Sayed, M.A.; El-Ganiny, A.M. Diclofenac mitigates virulence of multidrug-resistant Staphylococcus aureus. Microbiol. 2020, 202, 2751–2760.
- El-Ganiny, A.M.; Gad, A.I.; El-Sayed, M.A.; Shaldam, M.A.; Abbas, H.A. The promising anti-virulence activity of candesartan, domperidone, and miconazole on Staphylococcus aureus. J. Microbiol. 2021, 2021, 1–18. doi: 10.1007/s42770-021-00655-4
- Chen, F.; Di, H.; Wang, Y.; Cao, Q.; Xu, B.; Zhang, X.; Yang, N.; Liu, G.; Yang, C.-G.; Xu, Y.; et al. Small-molecule targeting of a diapophytoene desaturase inhibits aureus virulence. Nat. Chem. Biol. 2016, 12, 174–179.
- Sun, J.; Zhang, Y.; Su, J.; Dai, T.; Chen, J.; Zhang, L.; Wang, H.; Liu, W.; Huang, M.; Chen, Z. Naftifine enhances photodynamic therapy against Staphylococcus aureus by inhibiting staphyloxanthin expression. Pigm. 2020, 179, 108392.
- Wang, Y.; Chen, F.; Di, H.; Xu, Y.; Xiao, Q.; Wang, X.; Wei, H.; Lu, Y.; Zhang, L.; Zhu, J.J.J.O.M.C. Discovery of potent benzofuran-derived diapophytoene desaturase (CrtN) inhibitors with enhanced oral bioavailability for the treatment of methicillin-resistant Staphylococcus aureus (MRSA) infections. Med. Chem. 2016, 59, 3215–3230.
- Gao, P.; Davies, J.; Kao, R.Y.T. Dehydrosqualene desaturase as a novel target for anti-virulence therapy against Staphylococcus aureus. mBIO 2017, 8, e01224–e01217.
- Ni, S.; Li, B.; Chen, F.; Wei, H.; Mao, F.; Liu, Y.; Xu, Y.; Qiu, X.; Li, X.; Liu, J. Novel staphyloxanthin inhibitors with improved potency against multidrug resistant Staphylococcus aureus. ACS Med. Chem. Lett. 2018, 9, 233–237.
- Kong, E.F.; Tsui, C.; Kucharíková, S.; Andes, D.; van Dijck, P.; Jabra-Rizk, M.A. Commensal protection of Staphylococcus aureus against antimicrobials by Candida albicans biofilm matrix. mBio 2016, 7, e01365–e01316.
- Morales, D.K.; Hogan, D.A.J.P.P. Candida albicans interactions with bacteria in the context of human health and disease. PLoS Pathog. 2010, 6, e1000886.
- Vila, T.; Kong, E.F.; Ibrahim, A.; Piepenbrink, K.; Shetty, A.C.; McCracken, C.; Bruno, V.; Jabra-Rizk, M.A. Candida albicans quorum-sensing molecule farnesol modulates staphyloxanthin production and activates the thiol-based oxidative-stress response in Staphylococcus aureus. Virulence 2019, 10, 625–642.
- de Souza, L.S.; Puziol, L.C.; Tosta, C.L.; Bittencourt, M.L.; Santa Ardisson, J.; Kitagawa, R.R.; Filgueiras, P.R.; Kuster, R.M. Analytical methods to access the chemical composition of an Euphorbia tirucalli anticancer latex from traditional Brazilian medicine. Ethnopharmacol. 2019, 237, 255–265.
- Cui, N.; Zhang, L.; Quan, M.; Xu, J. Profile of the main bioactive compounds and in vitro biological activity of different solvent extracts from Ginkgo biloba exocarp. RSC Adv. 2020, 10, 45105–45111.
- Kowalczyk, M.; Golonko, A.; Świsłocka, R.; Kalinowska, M.; Parcheta, M.; Swiergiel, A.; Lewandowski, W. Drug design strategies for the treatment of viral disease. Plant phenolic compounds and their derivatives. Pharmacol. 2021, 12:709104. doi: 10.3389/fphar.2021.709104.
- Fatahala, S.S.; Khedr, M.A.; Mohamed, M.S. Synthesis and structure activity relationship of some indole derivatives as potential anti-inflammatory agents. Acta Chim. Slov. 2017, 64, 865–876.
- Tong, S.Y.C.; Sharma-Kuinkel, B.K.; Thaden, J.T.; Whitney, A.R.; Yang, S.-J.; Mishra, N.N.; Rude, T.; Lilliebridge, R.A.; Selim, M.A.; Ahn, S.H.; et al. Virulence of endemic nonpigmented northern Australian Staphylococcus aureus clone (clonal complex 75, argenteus) is not augmented by staphyloxanthin. J. Infect. Dis. 2013, 208, 520–527.
- Demuyser, L.; Jabra-Rizk, M.A.; Van Dijck, P. Microbial cell surface proteins and secreted metabolites involved in multispecies biofilms. Dis. 2014, 70, 219–230.
- Antonic, V.; Stojadinovic, A.; Zhang, B.; Izadjoo, M.J.; Alavi, M. Pseudomonas aeruginosa induces pigment production and enhances virulence in a white phenotypic variant of Staphylococcus aureus. Drug Resist. 2013, 6, 175.
- Ding, Y.; Liu, X.; Chen, F.; Di, H.; Xu, B.; Zhou, L.; Deng, X.; Wu, M.; Yang, C.-G.; Lan, L. Metabolic sensor governing bacterial virulence in Staphylococcus aureus. Natl. Acad. Sci. USA 2014, 111, E4981–E4990.
- Pandey, S.; Sahukhal, G.S.; Elasri, M.O. The msaABCR operon regulates the response to oxidative stress in Staphylococcus aureus. Bacteriol. Res. 2019, 201, e00417–e00419.
- Pannu, M.K.; Hudman, D.A.; Sargentini, N.J.; Singh, V.K. Role of SigB and staphyloxanthin in radiation survival of Staphylococcus aureus. Microbiol. 2019, 76, 70–77.
- García-Contreras, R.; Maeda, T.; Wood, T. Resistance to quorum-quenching compounds. Environ. Microbiol. 2013, 79, 6840–6846.
- Koch, B.; Liljefors, T.; Persson, T.; Nielsen, J.; Kjelleberg, S.; Givskov, M. The LuxR receptor: The sites of interaction with quorum-sensing signals and inhibitors. Microbiology 2005, 151, 3589–3602.
- Maura, D.; Ballok, A.E.; Rahme, L.G. Considerations and caveats in anti-virulence drug development. Opin. Microbiol. 2016, 33, 41–46.
- Ford, C.A.; Hurford, I.M.; Cassat, J.E. Antivirulence strategies for the treatment of Staphylococcus aureus infections: A mini review. Microbiol. 2021, 11:632706. doi: 10.3389/fmicb.2020.632706.
This entry is adapted from the peer-reviewed paper 10.3390/antibiotics11030298