Extracellular vesicles (EV) are a very diverse group of cell-derived vesicles released by almost all kind of living cells. EV are involved in intercellular exchange, both nearby and systemically, since they induce signals and transmit their cargo (proteins, lipids, miRNAs) to other cells, which subsequently trigger a wide variety of biological responses in the target cells. However, cell surface receptor-induced EV release is limited to cells from the immune system, including T lymphocytes. T cell receptor activation of T lymphocytes induces secretion of EV containing T cell receptors for antigen and several bioactive molecules, including proapoptotic proteins. These EV are specific for antigen-bearing cells, which make them ideal candidates for a cell-free, EV-dependent cancer therapy. In this review we examine the generation of EV by T lymphocytes and CAR-T cells and some potential therapeutic approaches of these EV.
- exosomes
- T lymphocytes
- immune synapse
- secretory granules
- multivesicular bodies
- cytotoxic activity
- cell death
- CAR T lymphocytes
1. Introduction
Extracellular Vesicle Types
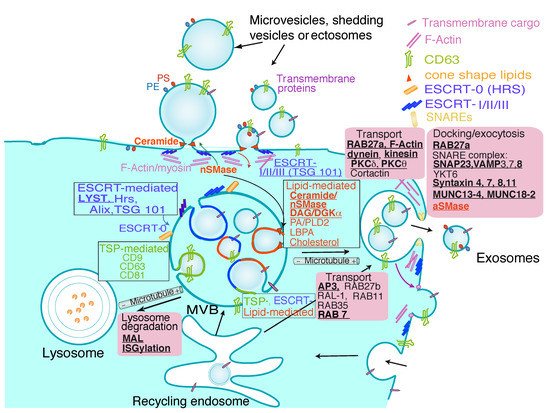
2. Extracellular Vesicles from T Lymphocytes
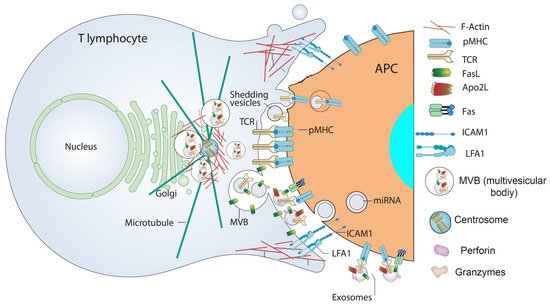
3. Chimeric Antigen Receptor (CAR) T Cells and CAR T Cell-Derived EV
Cancer Therapeutic Approaches
| Target Molecule | EV-Producing Cell | EV Types | EV Phenotype | Anti-Tumor Mechanism | Target Cell |
|---|---|---|---|---|---|
| EGFR, HER2 [55] |
Human CAR T cells (?) 1 |
Exosomes | CAR+, CD3+, CD63+, perforin+, granzyme B+, CD45−, CD28− |
Perforin/ granzyme B 2 |
EGFR+, HER2+ human breast cancer cells |
| HER2 [138] |
Human CAR T cells CD4+ (46%) CD8+ (49%) |
EV (small EV, probably exosomes plus larger EV) 3 |
CAR+, CD3+, CD63+, granzyme B+ |
Granzyme B 2 | HER2+ human breast cancer cells, ovarian cancer cells |
| Mesothelin [80] |
Human CAR T cells CD4+ (58%) CD8+ (31%) |
Probably exosomes 4 |
CAR+, CD3+, CD63+, perforin+, granzyme B+ |
Perforin/ granzyme B 2 |
Triple negative human breast cancer cells |
| CD19 [139] |
Human CAR HEK293 cells | Probably exosomes 4 |
CAR+, CD63+, CD81+ |
Indirect induction of proapoptotic genes in target cells |
CD19+ human B cell leukemia |
| CD19 [137] |
Human CAR HEK293 cells | Probably shedding vesicles 4 | CAR+, annexin V binding (PS exposure) |
MYC Gene disruption mediated by CRISPR/Cas9 |
CD19+ human B cell leukemia cell lines |
| Mesothelin CD19 [140] |
Human and mouse CAR T Cells (?) 1 |
EV 4 | Unknown 5 Contain RN7SL1 |
Recruitment of endogenous anti-tumor immunity byRN7SL1 |
Mouse melanoma expressing human CD19 |
| Event | CAR T Cells | CAR T Cell-Derived EV |
|---|---|---|
| Cytokine releasing syndrome | ++ | − |
| Neurotoxicity | ++ | − |
| Cross the blood barrier | − | ++ |
| Efficiency against solid tumors | +/− | ++ |
| Immunosuppression by tumoral PD-L1 | + | − |
| Immunological memory | + 1 | (?) 2 |
This entry is adapted from the peer-reviewed paper 10.3390/cells11050790