Coumarin compounds are attractive organic compounds with many practical applications. Among them there are compounds with biological activity, pharmaceuticals, agrochemicals, dyes, and optoelectronic materials. For this reason, enormous and continuous attempts were made to develop new synthetic pathways and protocols to facilitate the key cyclization reaction of heterocyclic ring and its regioselective functionalization. Of the numerous proposed reactions for the preparation of coumarins, those based on transition metal catalysts have been frequently used recently. Such processes as intramolecular and intermolecular hydroarylation of alkenes or alkynes can be mentioned among the most effective reactions proceeding via the activation of the C–H bond. Knowledge about the mechanistic foundations of catalytic processes seems to be of significant importance in order to improve them and simplify the conditions. Direct functionalization of the coumarin skeleton seems to be one of the more difficult tasks in recent times.
1. Introduction to Coumarins
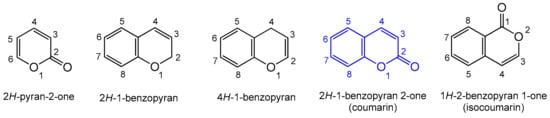
Heterocyclic compounds are an integral part of many biologically active molecules. Currently, many commercial drugs have a heterocyclic scaffold in their structure. Among them, bicyclic oxygen heterocycles resulting from fusion with the benzene ring constitute a large group of biologically active compounds (Figure 1).
Figure 1. Biologically important oxygen heterocycles.
The 2
H-pyran-2-one ring is commonly found in nature, and as a privileged biological scaffold, it exhibits a broad spectrum of actions including antifungal, antibiotic, and cytotoxic activities [
1,
2]. A condensed form of 2
H-pyran-2-one with the benzyl ring is known as coumarin. Coumarin is an organic heterocyclic compound belonging to the lactone subgroup with a benzo-α-pyrone (2
H-1-benzopyran-2-one) skeleton; the systematic nomenclature was established by the. IUPAC (International Union of Pure and Applied Chemistry) [
3]. Natural coumarin is obtained from tonka bean trees, which grow in Venezuela, Colombia, Guyana, and Brazil. The larger yields of coumarin can be obtained from the trees aged 7–10 years. Yellow–green fallen, mango-like fruits containing seeds are harvested from February to April. Pure coumarin is isolated by the alcoholic extraction of dry seeds or whole fruit.
Since the 19th century, coumarins have been a large class of natural compounds. It is estimated that more than 1300 natural coumarins isolated from the plants, fungi, and bacteria are known at present [
4]. In nature, coumarins are secondary metabolites and often play a protective role in inhibiting various biological processes. From a chemical point of view, they are molecules with several attractive features, such as a small molecular weight, simple structure, high bioavailability, good solubility in most organic solvents, and low toxicity. These features, together with the broad spectrum of biological activities, contribute to their significant role as leading compounds in the potential drugs study [
5,
6,
7].
The wide and diverse area of application for coumarins in medical sciences, biomedical research, and in many industries, including perfumery, is impressive. The pharmacological profile of these phytochemicals includes antibacterial (agasyllin, felamidin, and armillarisin A) [
8,
9], anti-inflammatory (coumarin and esculetin) [
10], anticancer [
11,
12,
13,
14], antiviral [
15,
16], antioxidant (Esculetin) [
17,
18,
19,
20], antifungal [
21], anticoagulant (warfarin) [
22,
23], anti-HIV (inophyllums and calanolides) [
24,
25,
26], and other activities [
27]. Coumarins also act as selective enzyme inhibitors; they are able to interact with targets in the treatment of diseases such as Parkinson’s and Alzheimer’s [
28,
29] and, more recently, they have been widely adopted in the design of small-molecule fluorescent chemosensors [
30].
2. Methods for the Synthesis of the Coumarin Core
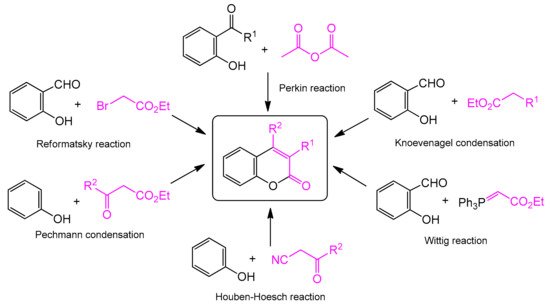
The classic and most frequently used one-step methods for the synthesis of coumarin derivatives include the Perkin reaction, von Pechmann condensation, Knoevenagel condensation, Baylis–Hillman reaction, Michael addition, Kostanecki reaction, and Heck lactonization reaction [
31] (
Scheme 1). The coumarin synthesis was first described by Perkin in 1868 [
32,
33]. The reaction consisted of heating sodium salt of salicylaldehyde with acetic anhydride. Further research into this process led to the preparation of cinnamic acid and its analogs using general synthesis that became known as the Perkin reaction. One of the most studied approaches to the synthesis of coumarins is the Pechmann reaction. It is based on the condensation of various phenols with β-ketoesters. It requires the presence of homogeneous catalysts, such as trifluoroacetic acid (TFA) [
34] or Lewis acids (LAs) (e.g., AlCl
3 [
35], ZnCl
2 [
36], ZrCl
4 [
37], and TiCl
4 [
38]) or heterogeneous catalysts such as cation exchange resins [
39], silica composites [
40], and zeolites [
41]. In the case of the Knoevenagel reaction, the transformation usually occurs via condensation of various
o-hydroxyaldehydes with active methylene compounds in the presence of a base catalyst [
42]. In turn, Houben–Hoesch condensation is based on the reaction of β-ketonitriles and phenol derivatives, and in the Wittig reaction, coumarin derivatives are formed as a result of the reaction of aromatic aldehydes or ketones with a phosphonate or phosphorous ylide.
Scheme 1. Traditional methods of coumarin synthesis.
The chemistry of transition metal complexes is widely applied in catalysis. The transition metals, united in the d-block, have an excellent ability to form coordination complexes consisting of a central metal atom and ligands. These complexes can induce numerous organic reactions, often under mild conditions and even in a stereoselective manner. For this reason, reactions in the presence of transition metals that are compatible with many functional groups have been successfully applied to the synthesis of coumarins.
3. C–H Bond Activation in Coumarin Synthesis
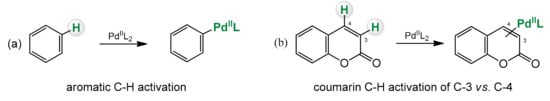
Organometallic reactions based on the activation of the C–H bond are a type of reactions where the strong C–H bond is broken and replaced with a C-X bond, where X can be anything else then H (
Scheme 2) [
52,
53].
Scheme 2. C–H bond activation of benzene (a) and coumarin (b).
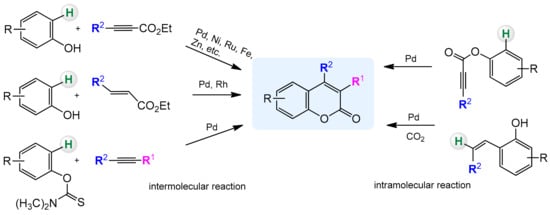
In recent years, C–H activation has become a versatile and effective tool in the preparation of heterocyclic compounds including coumarins and their derivatives. The traditional methods rely mainly on the condensation reactions of phenols with the carbonyl compounds and are catalyzed by a strong acid or base. Regioselectivity of these processes is not ideal and often leads to mixtures of compounds. Formation of C–C and C–O bonds in the presence of transition metal complexes via C–H activation is a good alternative to classical methods and partially solves this problem. In addition, it provides an efficient and cost-effective method for the direct synthesis of substituted coumarins from readily available arenes (
Scheme 3).
Scheme 3. Direct synthesis of coumarins by C–H bond activation (selected examples).
The coumarin backbone has six C–H sites and a C=C unit that are susceptible to functionalization. The aryl subunit of the coumarin ring system is not as reactive as in the case of the normal benzene derivative, and modifications in this region are quite difficult chemically, although possible by such reaction as C–H activation. The conjugated C=C bond between the C-3 and C-4 carbon is fixed in the
cis conformation and contributes to strong fluorescence emission and good photostability of coumarins. This bond is reactive and chemically available; hence, it is possible to obtain coumarins modified at C-3 and C-4 and even larger fused coumarin heterocyclic derivatives. The Kumada, Stille, Heck, Sonogashira, Negishi, Suzuki, or Buchwald–Hartwig coupling reactions are most often used for this purpose [
55,
56,
57,
58,
59,
60,
61,
62,
63]. The substrates in the abovementioned reactions are mostly coumarin derivatives such as vinyl coumarin, bromocoumarin, coumarin-3-carboxylic acid, or triflate-substituted coumarin. This is where the direct functionalization of the C–H bond emerges as an atom-economical and environmentally friendly synthetic tool that eliminates the need to prefunctionalize the coupling partners.
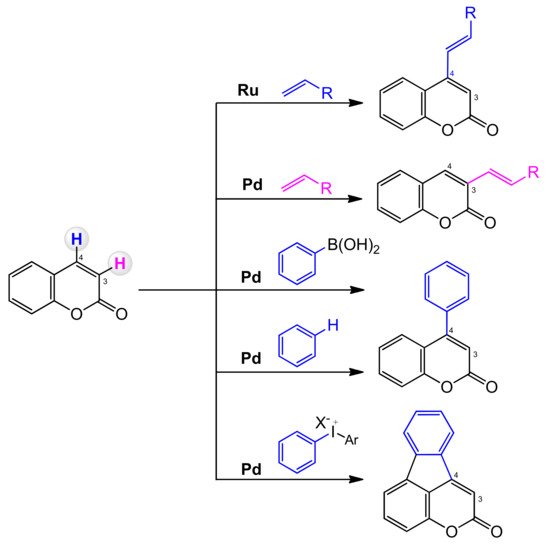
In coumarin, the two C–H bonds at C-3 and C-4 are chemically different. Direct activation of the C–H bond can sometimes be difficult and demanding. The C-3 position can undergo electrophilic palladation to form a C-3 palladium intermediate that allows regionally controlled arylation or alkenylation. It is also possible to predict the regioselectivity of the arylation and alkenylation reactions of the palladium catalyzed coumarins (
Scheme 4). The carbopalladation favors the delivery of the aryl group to the C-4 position, and the alkenylation process is usually C-3 selective [
64].
Scheme 4. Regioselectivity of coumarin C–H functionalization.
The selective functionalization of the C-4 position is more difficult, and its main limitation is the large dependence on palladium chemistry and the use of arenes as coupling partners. A solution to the problems of the selective functionalization of the C=C bond of coumarins is often the introduction of directing groups covalently tethered to the substrate at C-3 or C-4, which can coordinate to the transition metal center and direct the C–H activation process to the target site.
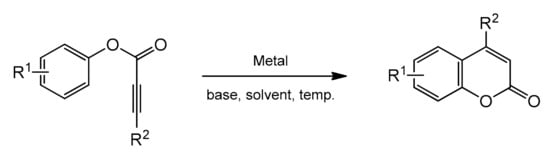
3.1. Direct Synthesis of Coumarins through Intermolecular Hydroarylation of Alkynes
Phenol and naphthol derivatives are proper substrates for the production of 4-arylcoumarins. The reaction of their C–H functionalization takes place in the ortho position, and the metals used as catalysts in this process are, first of all, palladium, iron, and platinum.
The palladium-catalyzed addition of phenols to alkynoates was initially demonstrated by Trost and co-workers [65,66]. The reactions proceeded with good yields at 35 °C with 10 mol% Pd(OAc)2 as a metal source and the addition of sodium acetate as a base and formic acid as a solvent was of key importance. Formic acid was proposed to influence the cyclization reaction by reducing PdII to Pd0. It was soon found that better results were achieved when using Pd2(dba)3 instead of Pd(OAc)2. The reaction proceeded effectively at room temperature with unsubstituted as well as alkyl- and aryl-substituted alkynoates (Table 1).
The Kitamura group presented the direct hydroarylation of alkynes using a Pd(OAc)2 catalyst in TFA/DCM [67,68,69]. ron-catalyzed hydroarylation offers an interesting and improved method for the synthesis of 4-substituted coumarins [71]. The reaction proceeded efficiently in the TFA/1,2-DCE solvent system. The yields reported with the iron catalyst, in some cases, were much higher than those obtained with palladium or platinum complexes.
3.2. Direct Synthesis of Coumarins through the Intramolecular Hydroarylation of Alkynes
Metal-catalyzed intramolecular hydroarylation of arylpropiolates via the activation of the C–H bond is an alternative one-step method resulting in theto coumarin preparation (
Scheme 7).
Scheme 7. Intramolecular hydroarylation of alkynes.
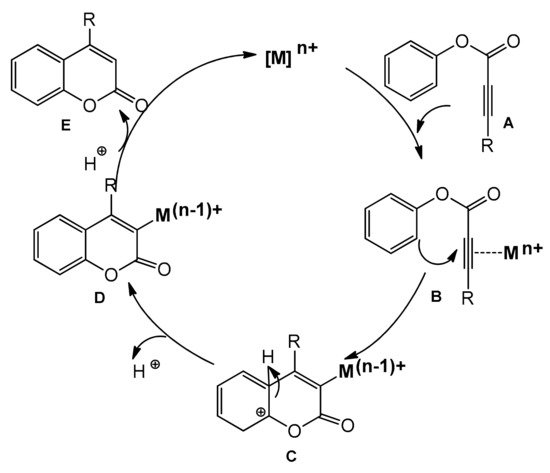
Recently, several improved versions of the reaction for the synthesis of 4-arylcoumarins with metal complexes as catalysts have been reported. In the course of the intramolecular hydroarylation process, the triple bond of
A is activated by the alkynophilic metal catalyst (Pd, Pt, Au), and the formed intermediate
B undergoes intramolecular cyclization leading to
C. The proton elimination produces the complex
D, which releases the coumarin derivative
E after the proto-demetallation and regenerates the catalyst (
Scheme 8).
Scheme 8. Intramolecular hydroarylation of C≡C.
3.3. Direct Synthesis of Coumarins via the Intermolecular Hydroarylation of Alkenes
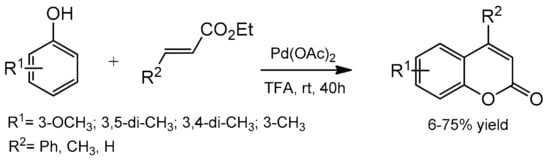
Direct C–H bond alkenylation provides another attractive strategy for coumarin synthesis, and some successful examples have been developed including Pd-catalyzed arylation of acrylates. The first palladium-catalyzed reaction was reported in 2005 by Kitamura and co-workers [
82]. The coumarin formation was accomplished at room temperature in TFA as the solvent and K
2S
2O
8 as the oxidant (
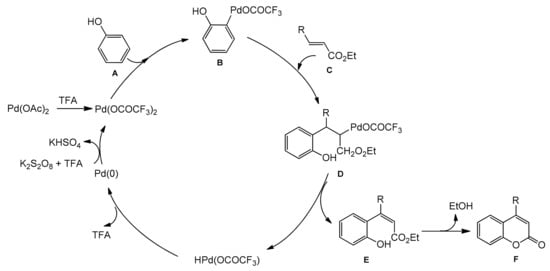
Scheme 9). The reaction of several phenols with ethyl cinnamate, ethyl crotonate, and ethyl acrylate gave the corresponding products in moderate to good yields. The oxidative coupling of electron-rich phenols was a competitive reaction under the reaction conditions thereby reducing product yields.
Scheme 9. Synthesis of coumarins from the phenol derivative and acrylates.
A possible reaction mechanism for the formation of coumarins is shown in
Scheme 10. It assumes that an arylpalladium species undergoes addition to the double bond of acrylate followed by the palladium hydride elimination instead of protonation by TFA. Therefore, an oxidant is needed to regenerate the palladium(II) moiety.
Scheme 10. Mechanism of Pd-catalyzed arylation of acrylates.
3.4. Direct Synthesis of Coumarins via Intramolecular Hydroarylation of Alkenes
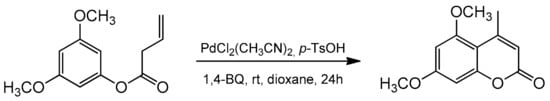
Recently, the Lete’ group presented the intramolecular Pd(II)-catalyzed alkenylation of aryl homoallyl ethers allowing access to substituted flavenes and isoflavenes [
86]. The reaction was conducted in the presence of PdCl
2(CH
3CN)
2 and 1,4-benzoquinone (1,4-BQ). The addition of
p-TsOH enhanced the system’s reactivity, and the synthesis could be performed in dioxane at room temperature for 24 h. The catalytic process was versatile, and the reaction condition also allowed for the synthesis of 5,7-dimethoxy-4-methylcoumarin (
Scheme 14).
Scheme 14. Synthesis of 5,7-dimethoxy-4-methylcoumarin.
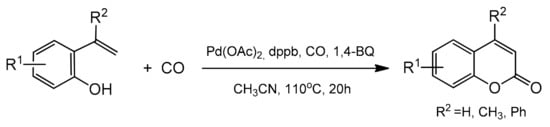
3.5. Intramolecular Cyclocarbonylation and Cyclocarboxylation
The cyclocarbonylation and cyclocarboxylation reactions have been demonstrated as a powerful route for the direct synthesis of the heterocyclic compounds [
88] including the coumarin skeleton. In 2012, a novel method for the synthesis of 4-methylcoumarin and 4-phenylcoumarin was developed by the Alper group [
89]. The reaction was based on the palladium-catalyzed oxidative cyclocarbonylation (
Scheme 16).
Scheme 16. Pd-catalyzed oxidative cyclocarbonylation of 2-vinylphenol.
3.6. Other Methods for Direct Synthesis of Coumarins by C–H Bond Activation
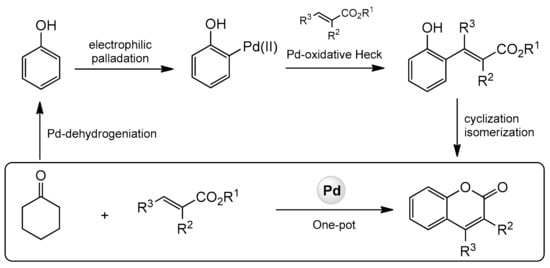
In 2013, the Hong group presented a very distinguished paper on the synthesis of C-3, C-4, and C-7-substituted coumarins [
93]. The reaction was catalyzed by palladium(II) using cyclohexanones and alkenes as substrates and consisted of a dehydrogenation-oxidative Heck-cyclization sequence (
Scheme 21).
Scheme 21. Sequence of reactions for construction of substituted coumarins.
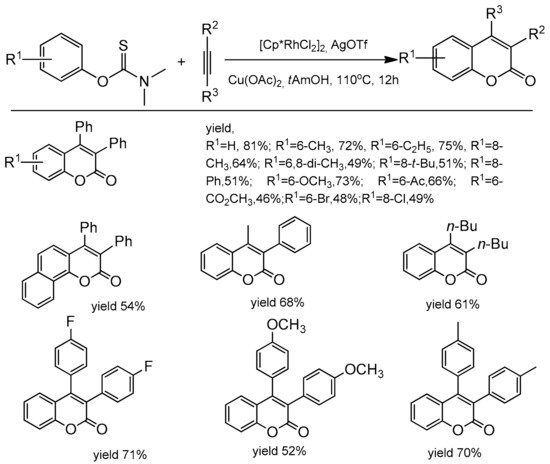
Xia and co-workers extended the access to C-3 and C-4 substituted coumarins making use of aryl thiocarbamates as the starting material [
94]. The reaction was catalyzed by [Cp*RhCl
2]
2 with a catalytic amount of AgOTf. Copper acetate was applied as an oxidant and
t-AmOH as a solvent. The reaction was heated for 12 h at 110 °C for complete conversion. In the reaction of diphenylacetylene with aryl thiocarbamates substituted at the
para position with the electron-donating groups the corresponding products were obtained with good yields (up to 82%). The same substituents at the
ortho position gave lower yields of products. The electron-deficient thiocarbamates also exhibited diminished reactivity. The process was compatible with ester, ketone, and halide functional groups (
Scheme 24). To obtain insight into the reaction mechanism, some control experiments and mechanism studies were performed and revealed that Cu(OAc)
2 acts both as the oxidant for Rh(I) and as the oxygen source for the carbonyl group.
Scheme 24. Rhodium-catalyzed reaction of aryl thiocarbamates with diarylacetylenes.
3.7. C–H bond Activation Strategy for the Synthesis of C-3 and C-4 Substituted Coumarins
3.7.1. C-3 Selective Reactions
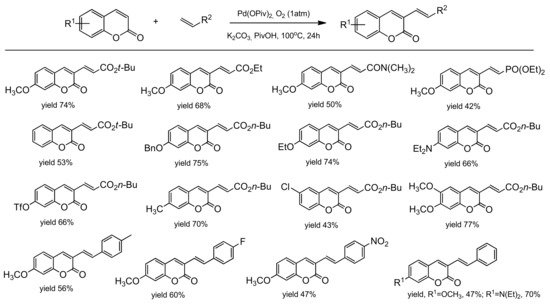
The palladium-catalyzed coupling reaction of coumarin with alkenes was explored by the Hong group [
96]. The desired coupling products were obtained using a catalytic system composed of Pd(OPiv)
2 and K
2CO
2 as a base. The C-3 alkenylation worked well in the open air and/or under 1 atm O
2 as the sole oxidant in PivOH at 100 °C. Various arylcoumarins with such functional groups as alkyl, chloro, benzoxy, methoxy, triflate, ethoxy as well as the coumarins substituted at C-3 with alkenes moiety, ester, phosphonate, and amide, were prepared in good yields. Their results are summarized in
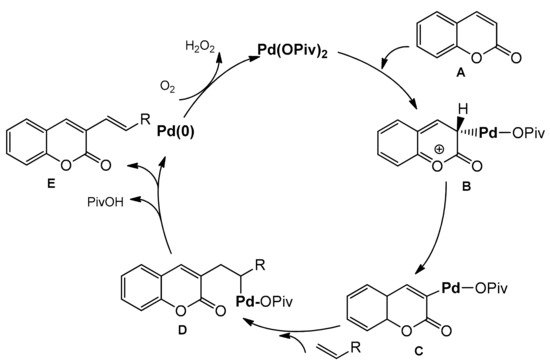
Scheme 25. To elucidate the alkenylation process, a mechanistic analysis was made leading to the proposed mechanism (
Scheme 26).
Scheme 25. Pd-catalyzed C-3 functionalization.
Scheme 26. Proposed mechanism of Pd-catalyzed C-3 functionalization.
3.7.2. C-4 Selective Reactions
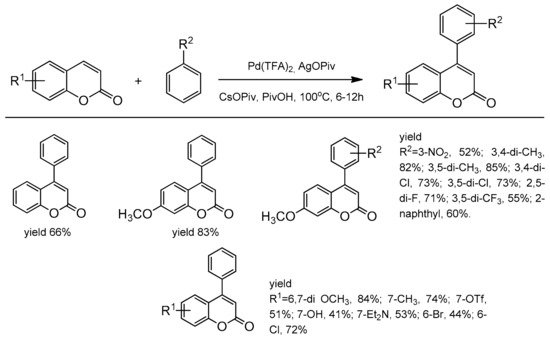
One of the first successful example of C-4 regiocontrolled C–H functionalization of coumarins was presented by Min and Hong [
102]. The simple non-activated arenes and coumarins participated in the Pd-catalyzed oxidative Heck coupling reaction. Among the palladium species, Pd(OPiv)
2 was the most effective in promoting coupling in the presence of AgOPiv and CsOPiv in pivalic acid at 100 °C. A series of 4-arylcoumarins (neoflavones) were obtained with very good yield and selectivity (
Scheme 33).
Scheme 33. C-4 selective arylation.
This entry is adapted from the peer-reviewed paper 10.3390/inorganics10020023