The chelating agent AAZTA features a mesocyclic seven-membered diazepane ring, conferring some of the properties of both acyclic and macrocyclic chelating agents. Described in the early 2000s, AAZTA and its derivatives exhibited interesting properties once complexed with metals and radiometals, combining a fast kinetic of formation with a slow kinetic of dissociation. Importantly, the extremely short coordination reaction times allowed by AAZTA derivatives were particularly suitable for short half-life radioelements (i.e., 68Ga).
- AAZTA
- bifunctional chelator
- radiopharmaceuticals
- theranostics
- nuclear medicine
1. Introduction
2. Design, Synthesis, and Kinetic Properties
2.1. Original AAZTA Derivatives
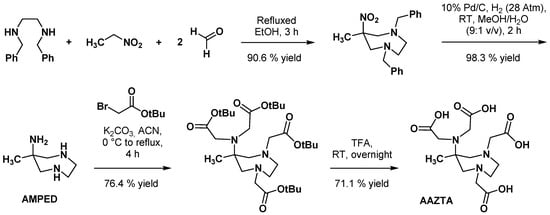
2.2. AAZTA Modulations to Improve Coordination Behavior
2.2.1. Modifications in the Number and Nature of Coordinating Side Arms
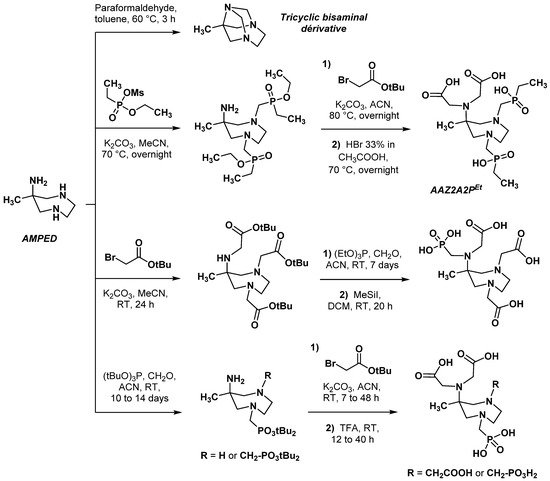
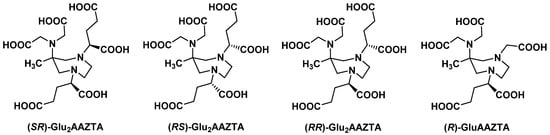
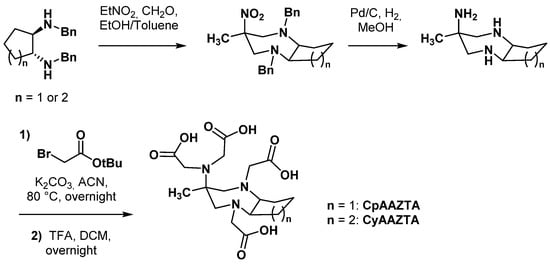
2.2.2. Backbone Rigidification
2.3. AAZTA Modulations to Obtain Bifunctional Chelating Agents (BCA)
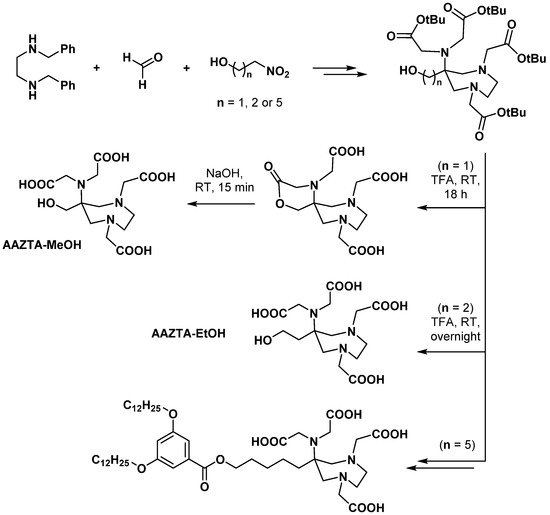
2.4. AAZ3A Derivatives
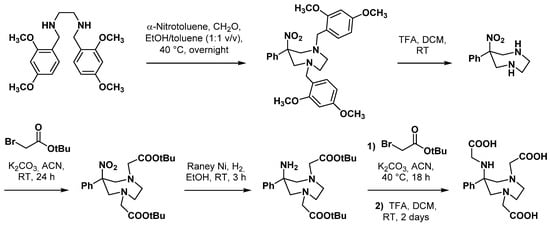
3. Applications in Nuclear Medicine
3.1. Monoclonal Antibodies
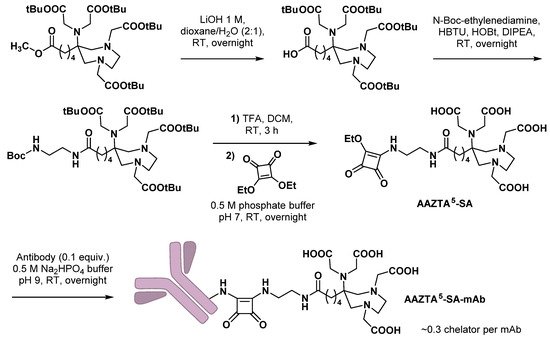
3.2. Small Molecules
3.2.1. Dual MRI/PET Imaging Agents
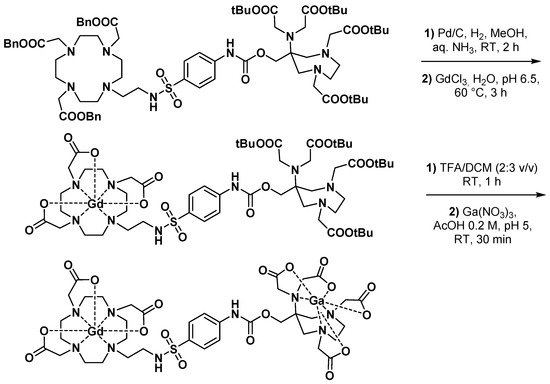
3.2.2. Bisphosphonate Derivatives
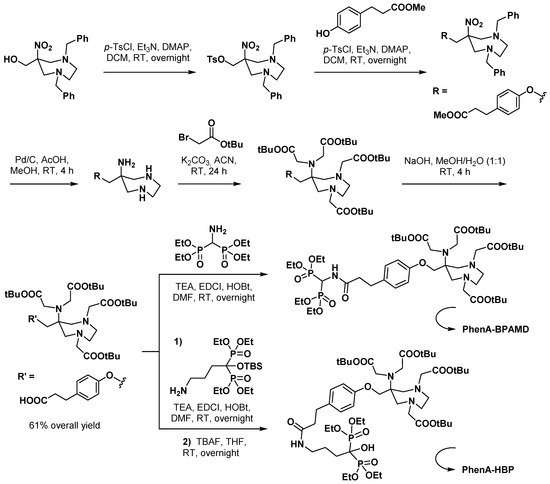
3.2.3. Curcumin Derivatives
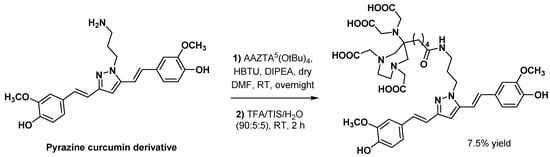
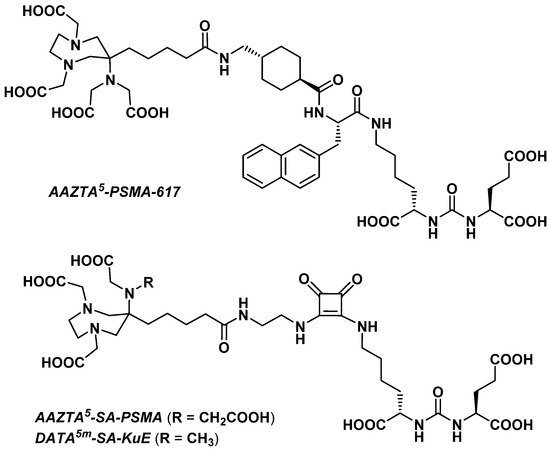
3.2.4. Prostate-Specific Antigen Ligands
Sinnes et al. explored the influence of the chelator part on the in vitro characteristics of a PSMA vector molecule by replacing DOTA with AAZTA5 in PSMA-617 [72]. The resulting AAZTA5-PSMA-617 (Figure 4) was successfully radiolabeled with 68Ga, 44Sc, and 177Lu at RT within 5 min, reaching >99% RCY for a chelator-to-radiometal molar ratio = 10:1. Both [68Ga]Ga- and [44Sc]Sc-AAZTA5-PSMA-617 were extremely stable in human serum, PBS, and EDTA/DTPA in PBS (>90% RCP over 2 h for the 68Ga derivative and >95% RCP over 8 h for the 44Sc derivative).
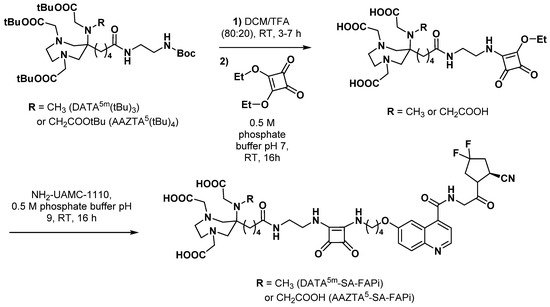
3.2.5. Fibroblast Activation Protein Inhibitors
3.3. Peptides
3.3.1. RGD Peptides
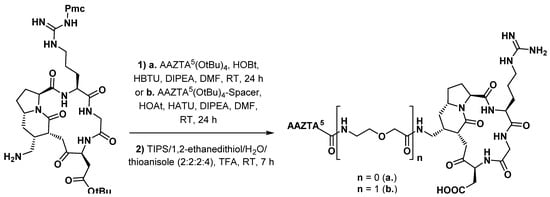
3.3.2. Gastrin Analogues
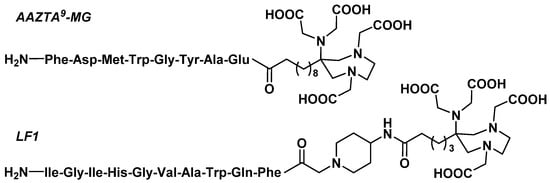
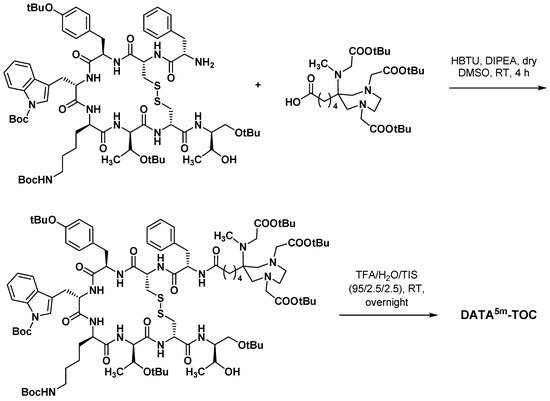
3.3.3. Octreotide Analogues
4. Conclusions
This entry is adapted from the peer-reviewed paper 10.3390/ph15020234