Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) is the causing pathogen of the unprecedented global Coronavirus Disease 19 (COVID-19) pandemic. Upon infection, the virus manipulates host cellular machinery and ribosomes to synthesize its own proteins for successful replication and to facilitate further infection. SARS-CoV-2 executes a multi-faceted hijacking of the host mRNA translation and cellular protein synthesis. Viral nonstructural proteins (NSPs) interact with a range of different ribosomal states and interfere with mRNA translation. Concurrent mutations on NSPs and spike proteins contribute to the epidemiological success of variants of concern (VOCs). The interactions between ribosomes and SARS-CoV-2 represent attractive targets for the development of antiviral therapeutics and vaccines.
- ribosome
- SARS-CoV-2
- COVID-19 mRNA vaccines
1. Introduction
2. SARS-CoV-2 Interfere with Ribosome mRNA Translation
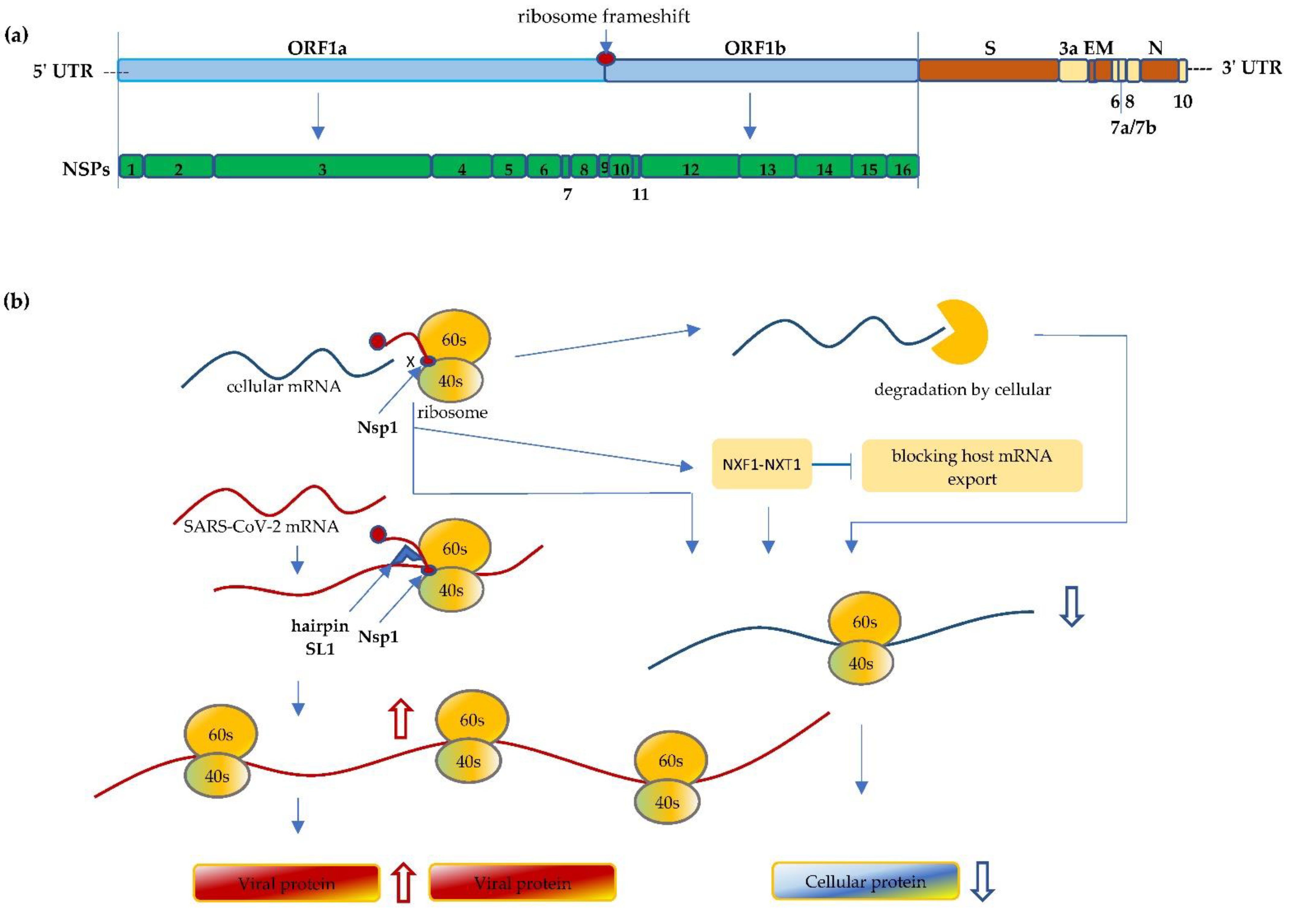
3. Mutations Impact Replication and Virulence
4. Molecule Design Impacts Ribosome Vaccine mRNA Translation
This entry is adapted from the peer-reviewed paper 10.3390/life12010057
References
- Zhou, P.; Yang, X.-L.; Wang, X.-G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.-R.; Zhu, Y.; Li, B.; Huang, C.-L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273.
- Zhu, N.; Zhang, D.; Wang, W.; Li, X.; Yang, B.; Song, J.; Zhao, X.; Huang, B.; Shi, W.; Lu, R.; et al. A Novel Coronavirus from Patients with Pneumonia in China. N. Engl. J. Med. 2020, 382, 727–733.
- WHO. Coronavirus Disease (COVID-1) Dashboard; WHO: Geneva, Switzerland, 2021.
- Nieto, B.; Gaspar, S.G.; Moriggi, G.; Pestov, D.G.; Bustelo, X.R.; Dosil, M. Identification of distinct maturation steps involved in human 40S ribosomal subunit biosynthesis. Nat. Commun. 2020, 11, 156.
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544.
- Chan, J.F.-W.; Kok, K.-H.; Zhu, Z.; Chu, H.; To, K.K.-W.; Yuan, S.; Yuen, K.-Y. Genomic characterization of the 2019 novel human-pathogenic coronavirus isolated from a patient with atypical pneumonia after visiting Wuhan. Emerg. Microbes Infect. 2020, 9, 221–236.
- Kim, D.; Kim, S.; Park, J.; Chang, H.R.; Chang, J.; Ahn, J.; Park, H.; Park, J.; Son, N.; Kang, G.; et al. A high-resolution temporal atlas of the SARS-CoV-2 translatome and transcriptome. Nat. Commun. 2021, 12, 5120.
- WHO. Tracking SARS-CoV-2 Variants; WHO: Geneva, Switzerland, 2021.
- Abdelghany, T.; Ganash, M.; Bakri, M.M.; Qanash, H.; Al-Rajhi, A.M.; Elhussieny, N.I. SARS-CoV-2, the other face to SARS-CoV and MERS-CoV: Future predictions. Biomed. J. 2021, 44, 86–93.
- Abdelrahman, Z.; Li, M.; Wang, X. Comparative Review of SARS-CoV-2, SARS-CoV, MERS-CoV, and Influenza A Respiratory Viruses. Front. Immunol. 2020, 11, 552909.
- Mariano, G.; Farthing, R.J.; Lale-Farjat, S.L.M.; Bergeron, J.R.C. Structural Characterization of SARS-CoV-2: Where We Are, and Where We Need to Be. Front. Mol. Biosci. 2020, 7, 605236.
- Finkel, Y.; Gluck, A.; Nachshon, A.; Winkler, R.; Fisher, T.; Rozman, B.; Mizrahi, O.; Lubelsky, Y.; Zuckerman, B.; Slobodin, B.; et al. SARS-CoV-2 uses a multipronged strategy to impede host protein synthesis. Nature 2021, 594, 240–245.
- Schubert, K.; Karousis, E.D.; Jomaa, A.; Scaiola, A.; Echeverria, B.; Gurzeler, L.-A.; Leibundgut, M.; Thiel, V.; Mühlemann, O.; Ban, N. SARS-CoV-2 Nsp1 binds the ribosomal mRNA channel to inhibit translation. Nat. Struct. Mol. Biol. 2020, 27, 959–966.
- Krichel, B.; Falke, S.; Hilgenfeld, R.; Redecke, L.; Uetrecht, C. Processing of the SARS-CoV pp1a/ab nsp7–10 region. Biochem. J. 2020, 477, 1009–1019.
- Bhatt, P.R.; Scaiola, A.; Loughran, G.; Leibundgut, M.; Kratzel, A.; Meurs, R.; Dreos, R.; O’Connor, K.M.; McMillan, A.; Bode, J.W.; et al. Structural basis of ribosomal frameshifting during translation of the SARS-CoV-2 RNA genome. Science 2021, 372, 1306–1313.
- Zimmer, M.M.; Kibe, A.; Rand, U.; Pekarek, L.; Ye, L.; Buck, S.; Smyth, R.P.; Cicin-Sain, L.; Caliskan, N. The short isoform of the host antiviral protein ZAP acts as an inhibitor of SARS-CoV-2 programmed ribosomal frameshifting. Nat. Commun. 2021, 12, 7193.
- Simeoni, M.; Cavinato, T.; Rodriguez, D.; Gatfield, D. I(nsp1)ecting SARS-CoV-2–ribosome interactions. Commun. Biol. 2021, 4, 715.
- Sun, Y.; Abriola, L.; Niederer, R.O.; Pedersen, S.F.; Alfajaro, M.M.; Monteiro, V.S.; Wilen, C.B.; Ho, Y.-C.; Gilbert, W.V.; Surovtseva, Y.V.; et al. Restriction of SARS-CoV-2 replication by targeting programmed −1 ribosomal frameshifting. Proc. Natl. Acad. Sci. USA 2021, 118, e2023051118.
- Lokugamage, K.G.; Narayanan, K.; Huang, C.; Makino, S. Severe Acute Respiratory Syndrome Coronavirus Protein nsp1 Is a Novel Eukaryotic Translation Inhibitor That Represses Multiple Steps of Translation Initiation. J. Virol. 2012, 86, 13598–13608.
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255.
- Baggen, J.; Vanstreels, E.; Jansen, S.; Daelemans, D. Cellular host factors for SARS-CoV-2 infection. Nat. Microbiol. 2021, 6, 1219–1232.
- Zhang, K.; Miorin, L.; Makio, T.; Dehghan, I.; Gao, S.; Xie, Y.; Zhong, H.; Esparza, M.; Kehrer, T.; Kumar, A.; et al. Nsp1 protein of SARS-CoV-2 disrupts the mRNA export machinery to inhibit host gene expression. Sci. Adv. 2021, 7, eabe7386.
- Min, Y.-Q.; Mo, Q.; Wang, J.; Deng, F.; Wang, H.; Ning, Y.-J. SARS-CoV-2 nsp1: Bioinformatics, Potential Structural and Functional Features, and Implications for Drug/Vaccine Designs. Front. Microbiol. 2020, 11, 587317.
- Xia, H.; Cao, Z.; Xie, X.; Zhang, X.; Chen, J.Y.-C.; Wang, H.; Menachery, V.D.; Rajsbaum, R.; Shi, P.-Y. Evasion of Type I Interferon by SARS-CoV-2. Cell Rep. 2020, 33, 108234.
- Tidu, A.; Janvier, A.; Schaeffer, L.; Sosnowski, P.; Kuhn, L.; Hammann, P.; Westhof, E.; Eriani, G.; Martin, F. The viral protein NSP1 acts as a ribosome gatekeeper for shutting down host translation and fostering SARS-CoV-2 translation. RNA 2021, 27, 253–264.
- Boehm, E.; Kronig, I.; Neher, R.A.; Eckerle, I.; Vetter, P.; Kaiser, L.; Geneva Centre for Emerging Viral Diseases. Novel SARS-CoV-2 variants: The pandemics within the pandemic. Clin. Microbiol. Infect. 2021, 27, 1109–1117.
- Davies, N.G.; Abbott, S.; Barnard, R.C.; Jarvis, C.I.; Kucharski, A.J.; Munday, J.D.; Pearson, C.A.B.; Russell, T.W.; Tully, D.C.; Washburne, A.D.; et al. Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England. Science 2021, 372, eabg3055.
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 variant of concern in South Africa. Nature 2021, 592, 438–443.
- Hoffmann, M.; Arora, P.; Groß, R.; Seidel, A.; Hörnich, B.F.; Hahn, A.S.; Krüger, N.; Graichen, L.; Hofmann-Winkler, H.; Kempf, A.; et al. SARS-CoV-2 variants B.1.351 and P.1 escape from neutralizing antibodies. Cell 2021, 184, 2384–2393.e12.
- Reardon, S. How the Delta variant achieves its ultrafast spread. Nat. Cell Biol. 2021, 21, 134.
- Scudellari, M. How the coronavirus infects cells- and why Delta is so dangerous. Nat. Cell Biol. 2021, 595, 640–644.
- CDC. SARS-CoV-2 Variant Classifications and Definitions. Available online: https://www.cdc.gov/coronavirus/2019-ncov/variants/variant-classifications.html#anchor_1632154493691 (accessed on 31 December 2021).
- Khateeb, J.; Li, Y.; Zhang, H. Emerging SARS-CoV-2 variants of concern and potential intervention approaches. Crit. Care 2021, 25, 244.
- Singh, J.; Samal, J.; Kumar, V.; Sharma, J.; Agrawal, U.; Ehtesham, N.; Sundar, D.; Rahman, S.; Hira, S.; Hasnain, S. Structure-Function Analyses of New SARS-CoV-2 Variants B.1.1.7, B.1.351 and B.1.1.28.1: Clinical, Diagnostic, Therapeutic and Public Health Implications. Viruses 2021, 13, 439.
- Sanches, P.R.; Charlie-Silva, I.; Braz, H.L.; Bittar, C.; Calmon, M.F.; Rahal, P.; Cilli, E.M. Recent advances in SARS-CoV-2 Spike protein and RBD mutations comparison between new variants Alpha (B.1.1.7, United Kingdom), Beta (B.1.351, South Africa), Gamma (P.1, Brazil) and Delta (B.1.617.2, India). J. Virus Erad. 2021, 7, 100054.
- Kannan, S.R.; Spratt, A.N.; Cohen, A.R.; Naqvi, S.H.; Chand, H.S.; Quinn, T.P.; Lorson, C.L.; Byrareddy, S.N.; Singh, K. Evolutionary analysis of the Delta and Delta Plus variants of the SARS-CoV-2 viruses. J. Autoimmun. 2021, 124, 102715.
- Ilmjärv, S.; Abdul, F.; Acosta-Gutiérrez, S.; Estarellas, C.; Galdadas, I.; Casimir, M.; Alessandrini, M.; Gervasio, F.L.; Krause, K.-H. Concurrent mutations in RNA-dependent RNA polymerase and spike protein emerged as the epidemiologically most successful SARS-CoV-2 variant. Sci. Rep. 2021, 11, 13705.
- FDA. Emergency Use Authorization (EUA) for Emergency Use of mRNA-1273. Available online: https://www.fda.gov/media/144636/download (accessed on 31 December 2021).
- FDA. Comirnaty (COVID-19 Vaccine, mRNA) Letter of Authorization. Available online: https://www.fda.gov/media/150386/download (accessed on 31 December 2021).
- Chaudhary, N.; Weissman, D.; Whitehead, K.A. mRNA vaccines for infectious diseases: Principles, delivery and clinical translation. Nat. Rev. Drug Discov. 2021, 20, 817–838.
- Walsh, E.E.; Frenck, R.W., Jr.; Falsey, A.R.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Neuzil, K.; Mulligan, M.J.; Bailey, R.; et al. Safety and Immunogenicity of Two RNA-Based COVID-19 Vaccine Candidates. N. Engl. J. Med. 2020, 383, 2439–2450.
- Chu, L.; McPhee, R.; Huang, W.; Bennett, H.; Pajon, R.; Nestorova, B.; Leav, B.; on behalf of the mRNA-1273 Study Group. A preliminary report of a randomized controlled phase 2 trial of the safety and immunogenicity of mRNA-1273 SARS-CoV-2 vaccine. Vaccine 2021, 39, 2791–2799.
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Marc, G.P.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA COVID-19 vaccine. N. Engl. J. Med. 2020, 383, 2603–2615.
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B.; et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N. Engl. J. Med. 2021, 384, 403–416.
- Corbett, K.S.; Edwards, D.K.; Leist, S.R.; Abiona, O.M.; Boyoglu-Barnum, S.; Gillespie, R.A.; Himansu, S.; Schäfer, A.; Ziwawo, C.T.; DiPiazza, A.T.; et al. SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness. Nature 2020, 586, 567–571.
- Svitkin, Y.V.; Cheng, Y.M.; Chakraborty, T.; Presnyak, V.; John, M.; Sonenberg, N. N1-methyl-pseudouridine in mRNA enhances translation through eIF2α-dependent and independent mechanisms by increasing ribosome density. Nucleic Acids Res. 2017, 45, 6023–6036.
- Wadhwa, A.; Aljabbari, A.; Lokras, A.; Foged, C.; Thakur, A. Opportunities and Challenges in the Delivery of mRNA-Based Vaccines. Pharmaceutics 2020, 12, 102.
- Ramanathan, A.; Robb, G.B.; Chan, S.-H. mRNA capping: Biological functions and applications. Nucleic Acids Res. 2016, 44, 7511–7526.
- Fechter, P.; Brownlee, G.G. Recognition of mRNA cap structures by viral and cellular proteins. J. Gen. Virol. 2005, 86, 1239–1249.
- Kozak, M. An analysis of vertebrate mRNA sequences: Intimations of translational control. J. Cell Biol. 1991, 115, 887–903.
- Pardi, N.; Hogan, M.J.; Weissman, D. Recent advances in mRNA vaccine technology. Curr. Opin. Immunol. 2020, 65, 14–20.
- Sahin, U.; Muik, A.; Vogler, I.; Derhovanessian, E.; Kranz, L.M.; Vormehr, M.; Quandt, J.; Bidmon, N.; Ulges, A.; Baum, A.; et al. BNT162b2 vaccine induces neutralizing antibodies and poly-specific T cells in humans. Nat. Cell Biol. 2021, 595, 572–577.
- Chatterjee, S.; Pal, J.K. Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol. Cell 2009, 101, 251–262.
- Berkovits, B.D.; Mayr, C. Alternative 3′ UTRs act as scaffolds to regulate membrane protein localization. Nat. Cell Biol. 2015, 522, 363–367.
- Jackson, N.A.C.; Kester, K.E.; Casimiro, D.; Gurunathan, S.; DeRosa, F. The promise of mRNA vaccines: A biotech and industrial perspective. NPJ Vaccines 2020, 5, 11.
- Weng, Y.; Li, C.; Yang, T.; Hu, B.; Zhang, M.; Guo, S.; Xiao, H.; Liang, X.-J.; Huang, Y. The challenge and prospect of mRNA therapeutics landscape. Biotechnol. Adv. 2020, 40, 107534.
- Xia, X. Detailed Dissection and Critical Evaluation of the Pfizer/BioNTech and Moderna mRNA Vaccines. Vaccines 2021, 9, 734.
- Weissman, D. mRNA transcript therapy. Expert Rev. Vaccines 2015, 14, 265–281.
- Trepotec, Z.; Aneja, M.K.; Geiger, J.; Hasenpusch, G.; Plank, C.; Rudolph, C. Maximizing the Translational Yield of mRNA Therapeutics by Minimizing 5′-UTRs. Tissue Eng. Part A 2019, 25, 69–79.
- Sample, P.J.; Wang, B.; Reid, D.W.; Presnyak, V.; McFadyen, I.J.; Morris, D.R.; Seelig, G. Human 5′ UTR design and variant effect prediction from a massively parallel translation assay. Nat. Biotechnol. 2019, 37, 803–809.
- Leppek, K.; Das, R.; Barna, M. Functional 5′ UTR mRNA structures in eukaryotic translation regulation and how to find them. Nat. Rev. Mol. Cell Biol. 2018, 19, 158–174.
- Ricci, E.; Limousin, T.; Rifo, R.S.; Rubilar, P.S.; Decimo, D.; Ohlmann, T. miRNA repression of translation in vitro takes place during 43S ribosomal scanning. Nucleic Acids Res. 2012, 41, 586–598.
- von Niessen, A.G.O.; Poleganov, M.A.; Rechner, C.; Plaschke, A.; Kranz, L.; Fesser, S.; Diken, M.; Löwer, M.; Vallazza, B.; Beissert, T.; et al. Improving mRNA-Based Therapeutic Gene Delivery by Expression-Augmenting 3′ UTRs Identified by Cellular Library Screening. Mol. Ther. 2019, 27, 824–836.
- Klauer, A.A.; Van Hoof, A. Degradation of mRNAs that lack a stop codon: A decade of nonstop progress. Wiley Interdiscip. Rev. RNA 2012, 3, 649–660.
- Jackson, R.J.; Hellen, C.U.T.; Pestova, T.V. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 2010, 11, 113–127.
- Hsieh, C.-L.; Goldsmith, J.A.; Schaub, J.M.; DiVenere, A.M.; Kuo, H.-C.; Javanmardi, K.; Le, K.C.; Wrapp, D.; Lee, A.G.; Liu, Y.; et al. Structure-based design of prefusion-stabilized SARS-CoV-2 spikes. Science 2020, 369, 1501–1505.
- Xia, X. Domains and Functions of Spike Protein in SARS-CoV-2 in the Context of Vaccine Design. Viruses 2021, 13, 109.
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immun. 2005, 23, 165–175.
- Roncati, L.; Corsi, L. Nucleoside-modified messenger RNA COVID-19 vaccine platform. J. Med Virol. 2021, 93, 4054–4057.
- Linares-Fernández, S.; Lacroix, C.; Exposito, J.-Y.; Verrier, B. Tailoring mRNA Vaccine to Balance Innate/Adaptive Immune Response. Trends Mol. Med. 2020, 26, 311–323.
- Xiong, Q.; Lee, G.Y.; Ding, J.; Li, W.; Shi, J. Biomedical applications of mRNA nanomedicine. Nano Res. 2018, 11, 5281–5309.
- Mugridge, J.S.; Coller, J.; Gross, J.D. Structural and molecular mechanisms for the control of eukaryotic 5′-3′ mRNA decay. Nat. Struct. Mol. Biol. 2018, 25, 1077–1085.
- Granados-Riveron, J.T.; Aquino-Jarquin, G. Engineering of the current nucleoside-modified mRNA-LNP vaccines against SARS-CoV-2. Biomed. Pharmacother. 2021, 142, 111953.