Approaches for effective and sustained drug delivery to the female reproductive tract (FRT) for treating a range of gynaecological conditions remain limited. The development of versatile delivery platforms, such as soluble gels (sol–gels) coupled with applicators/devices, holds considerable therapeutic potential for gynaecological conditions. Sol–gel systems, which undergo solution-to-gel transition, triggered by physiological conditions such as changes in temperature, pH, or ion composition, offer advantages of both solution- and gel-based drug formulations. Furthermore, they have potential to be used as a suitable drug delivery vehicle for other novel drug formulations, including micro- and nano-particulate systems, enabling the delivery of drug molecules of diverse physicochemical character. Hence, such systems are are of profound significance in delivering the drugs to various parts of FRT for optimal treatment of various gynecological conditions which was not achievable using conventional drug delivery technologies.
- vaginal drug delivery
- sol–gel formulations
- stimuli-responsive polymers
- mucoadhesion
- vaginal applicators/devices
1. Introduction
2. Sol–Gel Platform Technology in Vaginal Drug Delivery System
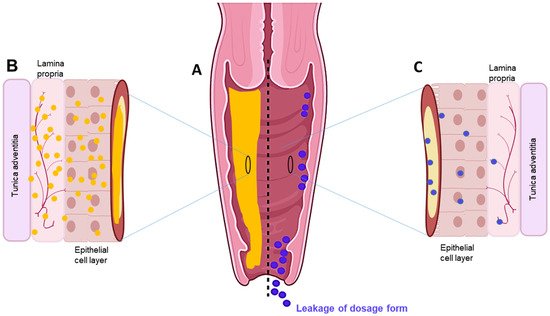
2.1. Features and Use of Vaginal Sol–Gel Formulations
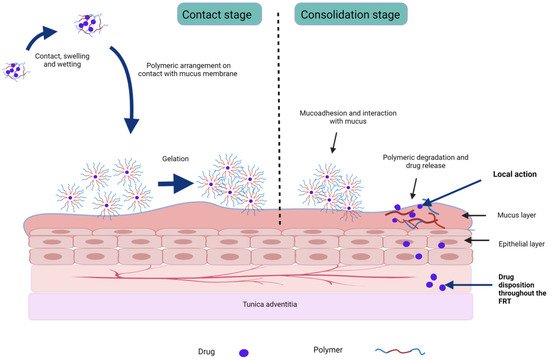
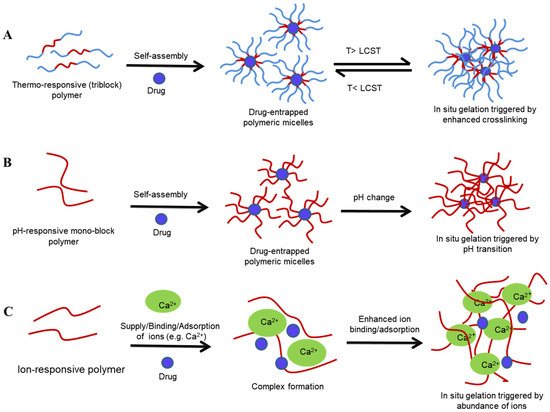
2.2. In Situ Sol-to-Gel Phase Transition Stimuli
2.2.1. Thermoresponsive Gelation
-
Poloxamers
-
Cellulose derivatives
-
Gelatin
2.2.2. pH Sensitive Sol–Gel Systems
-
Chitosan
-
Polyacrylates (PA)
2.2.3. Ion-Sensitive Sol–Gel Systems
-
Gellan gum
-
Alginate
-
Pectin
3. Applicators for Intravaginal Administration of Dosage Forms
| Applicator Type | Dimensions (mm) | Features | Advantages | Disadvantages | Product Examples | Reference |
|---|---|---|---|---|---|---|
| Single use | 114 × 12.7 with a tapered, rounded tip | Comprises plunger, barrel, and cap fabricated from PP and a piston inside the barrel made of non-latex rubber; pre-filled or manual filling |
Reduced cost due to bulk production | Higher plastic waste | KY-gel; Canesten® cream |
[217,218,219] |
| Multiple use | 114.5 × 11.3 | Comprises barrel and plunger fabricated from PE | Can be refilled and reusable, reducing packaging, storage, and transportation costs | Sanitary concerns | Ovestin® intravaginal cream | [215,218,219] |
| Single-use squeeze tube | 105 × 29 tube, plus 5-mm-wide applicator tip | Single-piece device fabricated from PE | Pre-filled, cost-effective | Cannot be filled manually |
Norden-Pac applicator |
[218] |
| Multiple pores | - | Presence of PE-fabricated membrane around the reservoir, infused with drug product and with perforations | Covers entire vaginal mucosa immediately after application; uniform drug delivery; pre-filled; biodegradable | High manufacturing cost | Universal vaginal applicator | [217] |
- Vigani, B.; Rossi, S.; Sandri, G.; Bonferoni, M.C.; Caramella, C.M.; Ferrari, F. Recent Advances in the Development of In Situ Gelling Drug Delivery Systems for Non-Parenteral Administration Routes. Pharmaceutics 2020, 12, 859.
- Pandey, M.; Choudhury, H.; Abdul-Aziz, A.; Bhattamisra, S.K.; Gorain, B.; Carine, T.; Wee Toong, T.; Yi, N.J.; Win Yi, L. Promising Drug Delivery Approaches to Treat Microbial Infections in the Vagina: A Recent Update. Polymers 2021, 13, 26.
- Neves, J.D.; de Oliveira, R.P.; de Oliveira, A.P.; Rodrigues, F.; Sarmento, B. Vaginal mucosa and drug delivery. In Mucoadhesive materials and Drug Delivery Systems, 1st edition; Wiley: Chichester, UK, 2014; pp. 99–132.
- Wong, T.W.; Dhanawat, M.; Rathbone, M.J. Vaginal drug delivery: Strategies and concerns in polymeric nanoparticle development. Expert Opin. Drug Deliv. 2014, 11, 1419–1434.
- Mirza, M.A.; Panda, A.K.; Asif, S.; Verma, D.; Talegaonkar, S.; Manzoor, N.; Khan, A.; Ahmed, F.J.; Dudeja, M.; Iqbal, Z. A vaginal drug delivery model. Drug Deliv. 2016, 23, 3123–3134.
- Cook, M.T.; Brown, M.B. Polymeric gels for intravaginal drug delivery. Control. Release 2018, 270, 145–157.
- Caramella, C.M.; Rossi, S.; Ferrari, F.; Bonferoni, M.C.; Sandri, G. Mucoadhesive and thermogelling systems for vaginal drug delivery. Drug Deliv. Rev. 2015, 92, 39–52.
- Matanović, M.R.; Kristl, J.; Grabnar, P.A. Thermoresponsive polymers: Insights into decisive hydrogel characteristics, mechanisms of gelation, and promising biomedical applications. J. Pharm. 2014, 472, 262–275.
- Ajazuddin; Alexander, A.; Khan, J.; Giri, T.K.; Tripathi, D.K.; Saraf, S.; Saraf, S. Advancement in stimuli triggered in situ gelling delivery for local and systemic route. Expert Opin. Drug Deliver. 2012, 9, 1573–1592.
- Jalalvandi, E.; Shavandi, A. In situ-forming and pH-responsive hydrogel based on chitosan for vaginal delivery of therapeutic agents. Mater. Sci. Mater. Med. 2018, 29, 1–11.
- Argenta, D.F.; Bernardo, B.D.C.; Chamorro, A.F.; Matos, P.R.; Caon, T. Thermosensitive hydrogels for vaginal delivery of secnidazole as an approach to overcome the systemic side-effects of oral preparations. J. Pharm. Sci. 2021, 159, 105722.
- Mennini, N.; Casella, G.; Cirri, M.; Maestrelli, F.; Mura, P. Development of cyclodextrin hydrogels for vaginal delivery of dehydroepiandrosterone. Pharm. Pharmacol. 2016, 68, 762–771.
- Date, A.A.; Shibata, A.; Goede, M.; Sanford, B.; la Bruzzo, K.; Belshan, M.; Destache, C.J. Development and evaluation of a thermosensitive vaginal gel containing raltegravir+ efavirenz loaded nanoparticles for HIV prophylaxis. Antiviral Res. 2012, 96, 430–436.
- Antimisiaris, S.G.; Mourtas, S. Recent advances on anti-HIV vaginal delivery systems development. Drug Deliv. Rev. 2015, 92, 123–145.
- Dorr, M.L.; Pierson, R.C.; Daggy, J.; Quinney, S.K.; Haas, D.M. Buccal versus vaginal misoprostol for term induction of labor: A retrospective cohort study. J. Perinatol. 2019, 36, 765.
- Tosti, C.; Biscione, A.; Morgante, G.; Bifulco, G.; Luisi, S.; Petraglia, F. Hormonal therapy for endometriosis: From molecular research to bedside. J. Obstet. Gynecol. Reprod. Biol. 2017, 209, 61–66.
- Elad, D.; Jaffa, A.J.; Grisaru, D. Biomechanics of early life in the female reproductive tract. Physiology 2020, 35, 134–143.
- Zierden, H.C.; Ortiz, J.I.; DeLong, K.; Yu, J.; Li, G.; Dimitrion, P.; Bensouda, S.; Laney, V.; Bailey, A.; Anders, N.M. Enhanced drug delivery to the reproductive tract using nanomedicine reveals therapeutic options for prevention of preterm birth. Transl. Med. 2021, 13.
- Das Neves, J.; Notario-Pérez, F.; Sarmento, B. Women-specific routes of administration for drugs: A critical overview. Drug Deliv. Rev. 2021, 176, 113865.
- Smoleński, M.; Karolewicz, B.; Gołkowska, A.M.; Nartowski, K.P.; Małolepsza-Jarmołowska, K. Emulsion-Based Multicompartment Vaginal Drug Carriers: From Nanoemulsions to Nanoemulgels. J. Mol. Sci. 2021, 22, 6455.
- Aplin, J. Uterus—Endometrium. In Encyclopedia of Reproduction, 2nd ed.; Skinner, M.K. Ed.; Academic Press: New York, NY, USA, 2018; pp. 326–332.
- Ellis, H. Anatomy of the uterus. Intensive Care Med. 2011, 12, 99–101.
- Myers, K.M.; Elad, D. Biomechanics of the human uterus. WIREs Syst. Biol. Med. 2017, 9, e1388.
- Friend, D.R. Drug delivery for the treatment of endometriosis and uterine fibroids. Drug Deliv. Transl. Res. 2017, 7, 829–839.
- Bahathiq, A.O.; Ledger, W.L. Historical Background and Functional Anatomy; Cambridge University: Cambridge, UK, 2010.
- Szmelskyj, I.; Aquilina, L.; Szmelskyj, A.O. Chapter 2—Anatomy and physiology of the reproductive system: Prerequirements for conception. An Integrated Approach to Treatment and Management ,In Acupuncture for IVF and Assisted Reproduction; Szmelskyj, I.; Aquilina, L.; Szmelskyj, A.O. Eds.; Churchill Livingstone: London, UK, 2015; pp. 23–58.
- Graziottin, A.; Gambini, D. Anatomy and physiology of genital organs—women. Clin. Neurol. 2015, 130, 39–60.
- Curlin, M.; Bursac, D. Cervical mucus: From biochemical structure to clinical implications. Biosci. 2013, 5, 507–515.
- Herfs, M.; Vargas, S.O.; Yamamoto, Y.; Howitt, B.E.; Nucci, M.R.; Hornick, J.L.; Mckeon, F.D.; Xian, W.; Crum, C.P. A novel blueprint for ‘top down’differentiation defines the cervical squamocolumnar junction during development, reproductive life, and neoplasia. Pathol. 2013, 229, 460–468.
- Yang, E.J.; Quick, M.C.; Hanamornroongruang, S.; Lai, K.; Doyle, L.A.; McKeon, F.D.; Xian, W.; Crum, C.P.; Herfs, M. Microanatomy of the cervical and anorectal squamocolumnar junctions: A proposed model for anatomical differences in HPV-related cancer risk. Pathol. 2015, 28, 994–1000.
- Taurin, S.; Almomen, A.A.; Pollak, T.; Kim, S.J.; Maxwell, J.; Peterson, C.M.; Owen, S.C.; Janát-Amsbury, M.M. Thermosensitive hydrogels a versatile concept adapted to vaginal drug delivery. Drug Target. 2018, 26, 533–550.
- Ng, K.Y.B.; Mingels, R.; Morgan, H.; Macklon, N.; Cheong, Y. In vivo oxygen, temperature and pH dynamics in the female reproductive tract and their importance in human conception: A systematic review. Reprod. Update 2018, 24, 15–34.
- Ding, N.; He, Y.; Qi, Y.; Zhang, H.; Xu, J.; Lei, J.; Yuan, L.; Ma, L.; Xue, H.; Jin, Z. Endometrial T2 values and thickness measured during the spontaneous menstrual cycle: Potential imaging biomarker related to female physiological hormones. J. Acad Radiol. 2021, 4, 1–7
- Lykke, M. R.; Becher, N.; Haahr, T.; Boedtkjer, E.; Jensen, J. S.; Uldbjerg, N., Vaginal, Cervical and Uterine pH in Women with Normal and Abnormal Vaginal Microbiota. Pathogens 2021, 10 (2), 90.
- Martyn, F.; McAuliffe, F.; Wingfield, M., The role of the cervix in fertility: is it time for a reappraisal? Reprod. 2014, 29 (10), 2092-2098.
- Taherali, F.; Varum, F.; Basit, A. W., A slippery slope: On the origin, role and physiology of mucus. Drug Deliv. Rev. 2018, 124, 16-33.
- Lalan, M. S.; Patel, V. N.; Misra, A., Polymers in vaginal drug delivery: Recent advancements. In Applications of Polymers in Drug Delivery, 2nd ed.; Elsevier: Amsterdam, The Netherlands, 2021; pp 281-303.
- Nakano, F. Y.; Leão, R. d. B. F.; Esteves, S. C., Insights into the role of cervical mucus and vaginal pH in unexplained infertility. MedicalExpress 2015, 2, 1-8.
- Justin-Temu, M.; Damian, F.; Kinget, R.; Mooter, G. V. D., Intravaginal gels as drug delivery systems. Womens Health 2004, 13 (7), 834-844.
- Muhleisen, A. L.; Herbst-Kralovetz, M. M., Menopause and the vaginal microbiome. Maturitas 2016, 91, 42-50.
- Godha, K.; Tucker, K.M.; Biehl, C.; Archer, D.F.; Mirkin, S. Human vaginal pH and microbiota: An update. Endocrinol. 2018, 34, 451–455.
- Clarke, M. A.; Rodriguez, A. C.; Gage, J. C.; Herrero, R.; Hildesheim, A.; Wacholder, S.; Burk, R.; Schiffman, M., A large, population-based study of age-related associations between vaginal pH and human papillomavirus infection. BMC Infect. Dis. 2012, 12 (1), 1-9.
- Klein, S.; Tietz, K., Vaginal and Intrauterine Delivery Systems. In In Vitro Drug Release Testing of Special Dosage Forms, John Wiley & Sons Ltd.: New York, USA, 2019; pp 177-209.
- Furst, T.; Piette, M.; Lechanteur, A.; Evrard, B.; Piel, G., Mucoadhesive cellulosic derivative sponges as drug delivery system for vaginal application. J. Pharm. Biopharm. 2015, 95, 128-135.
- Marcotte, H.; Krogh Andersen, K.; Lin, Y.; Zuo, F.; Zeng, Z.; Larsson, P. G.; Brandsborg, E.; Brønstad, G.; Hammarström, L., Characterization and complete genome sequences of L. rhamnosus DSM 14870 and L. gasseri DSM 14869 contained in the EcoVag® probiotic vaginal capsules. Res. 2017, 205, 88-98.
- Leyva-Gómez, G.; Prado-Audelo, D.; María, L.; Ortega-Peña, S.; Mendoza-Muñoz, N.; Urbán-Morlán, Z.; González-Torres, M.; Carmen, G.-D.; Figueroa-González, G.; Reyes-Hernández, O. D., Modifications in vaginal microbiota and their influence on drug release: challenges and opportunities. Pharmaceutics 2019, 11 (5), 217.
- Vigani, B.; Faccendini, A.; Rossi, S.; Sandri, G.; Bonferoni, M. C.; Grisoli, P.; Ferrari, F., Development of a mucoadhesive in situ gelling formulation for the delivery of Lactobacillus gasseri into vaginal cavity. Pharmaceutics 2019, 11 (10), 511.
- Kaambo, E.; Africa, C.; Chambuso, R.; Passmore, J.-A. S., Vaginal microbiomes associated with aerobic vaginitis and bacterial vaginosis. Front. Public Health 2018, 6, 78.
- Bernkop-Schnürch, A.; Hornof, M., Intravaginal Drug Delivery Systems: Design, Challenges, and Solutions. Am. J. Adv. Drug Deliv. 2003, 1 (4), 241-254.
- Osmałek, T.; Froelich, A.; Jadach, B.; Tatarek, A.; Gadziński, P.; Falana, A.; Gralińska, K.; Ekert, M.; Puri, V.; Wrotyńska-Barczyńska, J., Recent Advances in Polymer-Based Vaginal Drug Delivery Systems. Pharmaceutics 2021, 13 (6), 884.
- Hussain, A.; Ahsan, F., The vagina as a route for systemic drug delivery. J. Control. Release 2005, 103 (2), 301-313.
- Machado, R. M.; Palmeira-de-Oliveira, A.; Gaspar, C.; Martinez-de-Oliveira, J.; Palmeira-de-Oliveira, R., Studies and methodologies on vaginal drug permeation. Adv. Drug Deliv. Rev. 2015, 92, 14-26.
- Sassi, A. B.; McCullough, K. D.; Cost, M. R.; Hillier, S. L.; Rohan, L. C., Permeability of tritiated water through human cervical and vaginal tissue. J. Pharm. Sci. 2004, 93 (8), 2009-2016.
- Laksitorini, M.; Prasasty, V. D.; Kiptoo, P. K.; Siahaan, T. J., Pathways and progress in improving drug delivery through the intestinal mucosa and blood-brain barriers. Ther. Deliv. 2014, 5 (10), 1143-1163.
- Cicinelli, E.; Rubini, G.; De Ziegler, D.; Barba, B.; Pinto, V.; Di Stefano, M. G.; Mele, M., Absorption and preferential vagina-to-uterus distribution after vaginal administration of 99mTc-pertechnetate in postmenopausal women. Fertil. Steril. 2001, 76 (6), 1108-1112.
- Warren, M., Vaginal progesterone and the vaginal first-pass effect. Climacteric 2018, 21 (4), 355-357.
Patel, A.; Dhande, R.; Thakkar, H., Development of intravaginal rod insert bearing liposomal raloxifene hydrochloride and Leuprolide acetate as a potential carrier for uterine targeting. J. Pharm. Pharmacol. 2021, 73 (5), 653-663.
- das Neves, J.; Araújo, F.; Andrade, F.; Amiji, M.; Bahia, M. F.; Sarmento, B., Biodistribution and pharmacokinetics of dapivirine-loaded nanoparticles after vaginal delivery in mice. Pharm. Res. 2014, 31 (7), 1834-1845.
- Wang, X.; Liu, S.; Guan, Y.; Ding, J.; Ma, C.; Xie, Z., Vaginal drug delivery approaches for localized management of cervical cancer. Adv. Drug Deliv. Rev. 2021, 174, 114-126.
- Gomaa, E.; Lila, A. S. A.; Hasan, A. A.; Fakhr-eldin, S. G., Preparation and characterization of intravaginal vardenafil suppositories targeting a complementary treatment to boost in vitro fertilization process. Eur. J. Pharm. Sci. 2018, 111, 113-120.
- Lechanteur, A.; das Neves, J.; Sarmento, B., The role of mucus in cell-based models used to screen mucosal drug delivery. Adv. Drug Deliv. Rev. 2018, 124, 50-63.
- Vanić, Ž.; Škalko-Basnet, N., Nanopharmaceuticals for improved topical vaginal therapy: can they deliver? Eur. J. Pharm. Sci. 2013, 50 (1), 29-41.
- Sharma, P.; Srivastava, P.; Seth, A.; Tripathi, P. N.; Banerjee, A. G.; Shrivastava, S. K., Comprehensive review of mechanisms of pathogenesis involved in Alzheimer’s disease and potential therapeutic strategies. Prog. Neurobiol. 2019, 174, 53-89.
- Ensign, L. M.; Cone, R.; Hanes, J., Nanoparticle-based drug delivery to the vagina: a review. J. Control. Release 2014, 190, 500-514.
- Miller, E. A.; Beasley, D. E.; Dunn, R. R.; Archie, E. A., Lactobacilli dominance and vaginal pH: why is the human vaginal microbiome unique? Front. Microbiol. 2016, 7, 1936.
- Klatt, N. R.; Cheu, R.; Birse, K.; Zevin, A. S.; Perner, M.; Noël-Romas, L.; Grobler, A.; Westmacott, G.; Xie, I. Y.; Butler, J., Vaginal bacteria modify HIV tenofovir microbicide efficacy in African women. Science 2017, 356 (6341), 938-945.
- Xu, J.; Bian, G.; Zheng, M.; Lu, G.; Chan, W. Y.; Li, W.; Yang, K.; Chen, Z. J.; Du, Y., Fertility factors affect the vaginal microbiome in women of reproductive age. Am. J. Reprod. Immunol. 2020, 83 (4), e13220.
- Huang, B.; Fettweis, J. M.; Brooks, J. P.; Jefferson, K. K.; Buck, G. A., The changing landscape of the vaginal microbiome. Clin. Lab. Med. 2014, 34 (4), 747-761.
- Nayak, B. S.; Ellaiah, P.; Sudhahar, D., Novel approaches in vaginal drug delivery systems for local and systemic treatments. J. Pharm. Res. 2010, 3 (4), 675-80.
- Urbán-Morlán, Z.; Serrano-Mora, L. E.; Martínez-Acevedo, L.; Leyva-Gómez, G.; Mendoza-Muñoz, N.; Quintanar-Guerrero, D., New developments in intrauterine drug delivery systems and devices. In Drug Delivery Devices and Therapeutic Systems, Elsevier: Amsterdam, The Netherlands, 2021; pp 601-622.
- Aka-Any-Grah, A.; Bouchemal, K.; Koffi, A.; Agnely, F.; Zhang, M.; Djabourov, M.; Ponchel, G., Formulation of mucoadhesive vaginal hydrogels insensitive to dilution with vaginal fluids. Eur. J. Pharm. Biopharm. 2010, 76 (2), 296-303.
- Machado, R. M.; Palmeira-de-Oliveira, A.; Martinez-de-Oliveira, J.; Palmeira-de-Oliveira, R., Vaginal semisolid products: Technological performance considering physiologic parameters. Eur. J. Pharm. Sci. 2017, 109, 556-568.
- Sofi, H. S.; Abdal-Hay, A.; Ivanovski, S.; Zhang, Y. S.; Sheikh, F. A., Electrospun nanofibers for the delivery of active drugs through nasal, oral and vaginal mucosa: current status and future perspectives. Mater. Sci. Eng. C 2020, 111, 110756.
- Araújo, F.; Martins, C.; Azevedo, C.; Sarmento, B., Chemical modification of drug molecules as strategy to reduce interactions with mucus. Adv. Drug Deliv. Rev. 2018, 124, 98-106.
- Devadasu, V. R.; Deb, P. K.; Maheshwari, R.; Sharma, P.; Tekade, R. K., Physicochemical, pharmaceutical, and biological considerations in GIT absorption of drugs. In Dosage Form Design Considerations, Academic Press: Cambridge, MA, USA, 2018; pp 149-178.
- Mathias, N. R.; Hussain, M. A., Non-invasive Systemic Drug Delivery: Developability Considerations for Alternate Routes of Administration. J. Pharm. Sci. 2010, 99 (1), 1-20.
- Talegaonkar, S.; Iqbal, Z., In vitro/in vivo performance of different complexes of itraconazole used in the treatment of vaginal candidiasis. Braz. J. Pharm. Sci. 2012, 48 (4), 759-772.
- Jalalvandi, E.; Jafari, H.; Amorim, C. A.; Petri, D. F. S.; Nie, L.; Shavandi, A., Vaginal Administration of Contraceptives. Sci. Pharm. 2021, 89 (1), 3.
- Faisal, W.; Soliman, G. M.; Hamdan, A. M., Enhanced skin deposition and delivery of voriconazole using ethosomal preparations. J. Liposome Res. 2018, 28 (1), 14-21.
- Agrahari, V.; Putty, S.; Mathes, C.; Murowchick, J. B.; Youan, B. B. C., Evaluation of degradation kinetics and physicochemical stability of tenofovir. Drug Test. Anal. 2015, 7 (3), 207-213.
- Acharya, P. C.; Fernandes, C.; Suares, D.; Shetty, S.; Tekade, R. K., Solubility and solubilization approaches in pharmaceutical product development. In Dosage Form Design Considerations, Elsevier: Amsterdam, The Netherlands, 2018; pp 513-547.
- Passos, J. S.; Martino, L. C. d.; Dartora, V. F. C.; Araujo, G. L. B. d.; Ishida, K.; Lopes, L. B., Development, skin targeting and antifungal efficacy of topical lipid nanoparticles containing itraconazole. Eur. J. Pharm. Sci. 2020, 149, 105296.
- Van Eyk, A.; Van der Bijl, P.; Moll, L., Physicochemical characteristics of molecules and their diffusion across human vaginal mucosa. Eur. J. Inflamm. 2008, 6 (2), 65-71.
- Major, I.; McConville, C., Vaginal drug delivery for the localised treatment of cervical cancer. Drug Deliv. Transl. Res. 2017, 7 (6), 817-828.
- Ensign, L. M.; Hoen, T. E.; Maisel, K.; Cone, R. A.; Hanes, J. S., Enhanced vaginal drug delivery through the use of hypotonic formulations that induce fluid uptake. Biomaterials 2013, 34 (28), 6922-6929.
- Tomás, M.; Palmeira-de-Oliveira, A.; Simões, S.; Martinez-de-Oliveira, J.; Palmeira-de-Oliveira, R., Bacterial vaginosis: Standard treatments and alternative strategies. Int. J. Pharm. 2020, 587, 119659.
- Stewart, L. M.; Holman, C. D. A. J.; Hart, R.; Bulsara, M. K.; Preen, D. B.; Finn, J. C., In vitro fertilization and breast cancer: is there cause for concern? Fertil. Steril. 2012, 98 (2), 334-340.
- Liu, J. H.; Bernick, B.; Mirkin, S., Estradiol softgel inserts for the treatment of VVA symptoms: an expert opinion. Expert Opin. Drug Deliv. 2020, 17 (11), 1573-1581.
- Johal, H. S.; Garg, T.; Rath, G.; Goyal, A. K., Advanced topical drug delivery system for the management of vaginal candidiasis. Drug Deliv. 2016, 23 (2), 550-563.
- Shaker, D. S.; Ismail, S.; Hamed, S.; El-Shishtawy, E. M., Butoconazole nitrate vaginal sponge: Drug release and antifungal efficacy. J Drug Deliv Sci Technol. 2018, 48, 274-287.
- Ravel, J.; Moreno, I.; Simón, C., Bacterial vaginosis and its association with infertility, endometritis, and pelvic inflammatory disease. Am. J. Obstet. Gynecol. 2021, 224 (3), 251-257.
- Simões, A.; Veiga, F.; Vitorino, C.; Figueiras, A., A Tutorial for Developing a Topical Cream Formulation Based on the Quality by Design Approach. J. Pharm. Sci. 2018, 107 (10), 2653-2662.
- Tiboni, M.; Campana, R.; Frangipani, E.; Casettari, L., 3D printed clotrimazole intravaginal ring for the treatment of recurrent vaginal candidiasis. Int. J. Pharm. 2021, 596, 120290.
- Brache, V.; Faundes, A., Contraceptive vaginal rings: a review. Contraception 2010, 82 (5), 418-427.
- Lalan, M. S.; Patel, V. N.; Misra, A., Chapter 10 - Polymers in Vaginal Drug Delivery: Recent Advancements. In Applications of Polymers in Drug Delivery, 2nd ed.; Misra, A.; Shahiwala, A., Eds. Elsevier: Amsterdam, The Netherlands, 2021; pp 281-303.
- Iqbal, Z.; Dilnawaz, F., Nanocarriers for vaginal drug delivery. Recent patents on drug delivery & formulation 2019, 13 (1), 3-15.
- Machado, R. M.; Palmeira-De-Oliveira, A.; Martinez-De-Oliveira, J.; Palmeira-De-Oliveira, R., Vaginal Films for Drug Delivery. J. Pharm. Sci. 2013, 102 (7), 2069-2081.
- Palmeira-de-Oliveira, R.; Palmeira-de-Oliveira, A.; Martinez-de-Oliveira, J., New strategies for local treatment of vaginal infections. Adv. Drug Deliv. Rev. 2015, 92, 105-122.
- Coudray, M. S.; Madhivanan, P., Bacterial vaginosis—A brief synopsis of the literature. Eur. J. Obstet. Gynecol. Reprod. Biol. 2020, 245, 143-148.
- Vazquez, F.; Fernández-Blázquez, A.; García, B., Vaginosis. Vaginal microbiota. Enfermedades infecciosas y microbiologia clinica (English ed.) 2019, 37 (9), 592-601.
- Denning, D. W.; Kneale, M.; Sobel, J. D.; Rautemaa-Richardson, R., Global burden of recurrent vulvovaginal candidiasis: a systematic review. Lancet Infect Dis. 2018, 18 (11), e339-e347.
- Haas, D. M.; Daggy, J.; Flannery, K. M.; Dorr, M. L.; Bonsack, C.; Bhamidipalli, S. S.; Pierson, R. C.; Lathrop, A.; Towns, R.; Ngo, N.; Head, A.; Morgan, S.; Quinney, S. K., A comparison of vaginal versus buccal misoprostol for cervical ripening in women for labor induction at term (the IMPROVE trial): a triple-masked randomized controlled trial. Am. J. Obstet. Gynecol. 2019, 221 (3), 259.e1-259.e16.
- Pierce, S.; Bakker, R.; Myers, D. A.; Edwards, R. K., Clinical insights for cervical ripening and labor induction using prostaglandins. AJP Rep. 2018, 8 (4), e307.
- Gomez, H. B.; Hoffman, M. K.; Caplan, R.; Ruhstaller, K.; Young, M. H. H.; Sciscione, A. C., Buccal vs vaginal misoprostol combined with Foley catheter for cervical ripening at term (the BEGIN trial): a randomized controlled trial. Am. J. Obstet. Gynecol. 2021, 224 (5), 524.e1-524.e8.
- Abdelaziz, A.; Mahmoud, A. A.; Ellaithy, M. I.; Abees, S. H., Pre-induction cervical ripening using two different dinoprostone vaginal preparations: A randomized clinical trial of tablets and slow release retrievable insert. Taiwan J Obstet Gynecol. 2018, 57 (4), 560-566.
- Global HIV & AIDS statistics — 2020 fact sheet. https://www.unaids.org/en/resources/fact-sheet (accessed 05-28-2021).
- Traore, Y. L.; Chen, Y.; Ho, E. A., Current state of microbicide development. Clin. Pharmacol. Ther. 2018, 104 (6), 1074-1081.
- Coutinho, C.; Sarmento, B.; das Neves, J., Targeted microbicides for preventing sexual HIV transmission. J. Control. Release 2017, 266, 119-128.
- Mahalingam, A.; Jay, J. I.; Langheinrich, K.; Shukair, S.; McRaven, M. D.; Rohan, L. C.; Herold, B. C.; Hope, T. J.; Kiser, P. F., Inhibition of the transport of HIV in vitro using a pH-responsive synthetic mucin-like polymer system. Biomaterials 2011, 32 (33), 8343-8355.
- Fernández-Romero, J. A.; Teleshova, N.; Zydowsky, T. M.; Robbiani, M., Preclinical assessments of vaginal microbicide candidate safety and efficacy. Adv. Drug Deliv. Rev. 2015, 92, 27-38.
- Alexander, N. J.; Baker, E.; Kaptein, M.; Karck, U.; Miller, L.; Zampaglione, E., Why consider vaginal drug administration? Fertil. Steril. 2004, 82 (1), 1-12.
- Tai, F.-W.; Chang, C. Y.-Y.; Chiang, J.-H.; Lin, W.-C.; Wan, L., Association of pelvic inflammatory disease with risk of endometriosis: a nationwide cohort study involving 141,460 individuals. J. Clin. Med. 2018, 7 (11), 379.
- Yagur, Y.; Weitzner, O.; Tiosano, L. B.; Paitan, Y.; Katzir, M.; Schonman, R.; Klein, Z.; Miller, N., Characteristics of Pelvic Inflammatory Disease caused by Sexually transmitted disease–An epidemiologic Study. J. Gynecol. Obstet. Hum. Reprod. 2021, 50,102176.
- Falconer, H.; Yin, L.; Salehi, S.; Altman, D., Association between pelvic inflammatory disease and subsequent salpingectomy on the risk for ovarian cancer. Eur. J. Cancer 2021, 145, 38-43.
- Pathak, M.; Coombes, A. G. A.; Ryu, B.; Cabot, P. J.; Turner, M. S.; Palmer, C.; Wang, D.; Steadman, K. J., Sustained Simultaneous Delivery of Metronidazole and Doxycycline From Polycaprolactone Matrices Designed for Intravaginal Treatment of Pelvic Inflammatory Disease. J. Pharm. Sci. 2018, 107 (3), 863-869.
- Taylor, H. S.; Kotlyar, A. M.; Flores, V. A., Endometriosis is a chronic systemic disease: clinical challenges and novel innovations. Lancet 2021, 397 (10276), 839-852.
- Garzon, S.; Laganà, A. S.; Barra, F.; Casarin, J.; Cromi, A.; Raffaelli, R.; Uccella, S.; Franchi, M.; Ghezzi, F.; Ferrero, S., Novel drug delivery methods for improving efficacy of endometriosis treatments. Expert Opin. Drug Deliv. 2021, 18 (3), 355-367.
- Cicinelli, E., Intravaginal oestrogen and progestin administration: advantages and disadvantages. Best Pract Res Clin Obstet Gynaecol. 2008, 22 (2), 391-405.
- Godin, R.; Marcoux, V., Vaginally Administered Danazol: An Overlooked Option in the Treatment of Rectovaginal Endometriosis? J Obstet Gynaecol Can. 2015, 37 (12), 1098-1103.
- Mirkin, S.; Simon, J. A.; Liu, J. H.; Archer, D. F.; Castro, P. D.; Graham, S.; Bernick, B.; Komm, B., Evaluation of endometrial progesterone receptor expression after 12 weeks of exposure to a low-dose vaginal estradiol insert. Menopause 2021, 28 (9), 998.
- Casado-Espada, N. M.; de Alarcón, R.; de la Iglesia-Larrad, J. I.; Bote-Bonaechea, B.; Montejo, Á. L., Hormonal contraceptives, female sexual dysfunction, and managing strategies: a review. J. Clin. Med. 2019, 8 (6), 908.
- Sivasankaran, S.; Jonnalagadda, S., Advances in controlled release hormonal technologies for contraception: A review of existing devices, underlying mechanisms, and future directions. J. Control. Release 2021, 330, 797-811.
- Bahamondes, L.; Bahamondes, M. V., New and emerging contraceptives: a state-of-the-art review. Int J Womens Health 2014, 6, 221.
- Coppola, J. S., A New Vaginal pH Regulator for Hormone-Free, On-Demand Contraception. Nurs. Womens Health 2021, 25 (2), 152-155.
- Zhao, Y.-x.; Chen, S.-r.; Su, P.-p.; Huang, F.-h.; Shi, Y.-c.; Shi, Q.-y.; Lin, S., Using mesenchymal stem cells to treat female infertility: an update on female reproductive diseases. Stem Cells Int. 2019, 2019, 9071720.
- Christen, M.; Schertz, J. C.; Arriagada, P.; Keitel, J.; Müller, H., The redesigned follitropin α pen injector for infertility treatment. Expert Opin. Drug Deliv. 2011, 8 (6), 833-839.
- Saini, N.; Sodhi, R. K.; Bajaj, L.; Pandey, R. S.; Jain, U. K.; Katare, O. P.; Madan, J., Intravaginal administration of metformin hydrochloride loaded cationic niosomes amalgamated with thermosensitive gel for the treatment of polycystic ovary syndrome: In vitro and in vivo studies. Colloids Surf. B 2016, 144, 161-169.
- Sam, S.; Ehrmann, D. A., Metformin therapy for the reproductive and metabolic consequences of polycystic ovary syndrome. Diabetologia 2017, 60 (9), 1656-1661.
- Weiss, J. M.; Tauchert, S.; Ludwig, A. K.; Diedrich, K., Treatment strategies in PCOS patients. Reprod. Biomed. Online 2005, 10, 67-74.
- Briden, L.; Shirin, S.; Prior, J. C., The central role of ovulatory disturbances in the etiology of androgenic polycystic ovary syndrome (PCOS)—Evidence for treatment with cyclic progesterone. Drug Discov. Today Dis. Models 2020, 32, 71–82.
- Federico, C.; Sun, J.; Muz, B.; Alhallak, K.; Cosper, P. F.; Muhammad, N.; Jeske, A.; Hinger, A.; Markovina, S.; Grigsby, P., Localized delivery of cisplatin to cervical cancer improves its therapeutic efficacy and minimizes its side effect profile. Int. J. Radiat. Oncol. Biol. Phys. 2021, 109 (5), 1483-1494.
- Pickar, J.; Amadio, J.; Bernick, B.; Mirkin, S., Pharmacokinetic studies of solubilized estradiol given vaginally in a novel softgel capsule. Climacteric 2016, 19 (2), 181-187.
- Bolla, D.; Weissleder, S. V.; Radan, A. P.; Gasparri, M. L.; Raio, L.; Müller, M.; Surbek, D., Misoprostol vaginal insert versus misoprostol vaginal tablets for the induction of labour: a cohort study. BMC Pregnancy Childbirth 2018, 18 (1), 149.
- Peet, M. M.; Agrahari, V.; Anderson, S. M.; Hanif, H.; Singh, O. N.; Thurman, A. R.; Doncel, G. F.; Clark, M. R., Topical Inserts: A Versatile Delivery Form for HIV Prevention. Pharmaceutics 2019, 11 (8), 374.
- Küng, E.; Fürnkranz, U.; Walochnik, J., Chemotherapeutic options for the treatment of human trichomoniasis. Int. J. Antimicrob. Agents 2019, 53 (2), 116-127.
- Velázquez, N. S.; Turino, L. N.; Luna, J. A.; Mengatto, L. N., Progesterone loaded thermosensitive hydrogel for vaginal application: Formulation and in vitro comparison with commercial product. Saudi Pharm J. 2019, 27 (8), 1096-1106.
- Child, T.; Leonard, S. A.; Evans, J. S.; Lass, A., Systematic review of the clinical efficacy of vaginal progesterone for luteal phase support in assisted reproductive technology cycles. Reprod. Biomed. Online 2018, 36 (6), 630-645.
- Notario-Pérez, F.; Cazorla-Luna, R.; Martín-Illana, A.; Ruiz-Caro, R.; Tamayo, A.; Rubio, J.; Veiga, M.-D., Optimization of tenofovir release from mucoadhesive vaginal tablets by polymer combination to prevent sexual transmission of HIV. Carbohydr. Polym. 2018, 179, 305-316.
- Simon, J. A.; Pickar, J. H.; Shadiack, A. M.; Warrier, B.; Graham, S.; Bernick, B.; Mirkin, S., Physical characteristics and properties of estradiol softgel vaginal inserts. Menopause 2020, 27 (2), 150.
- Blakney, A. K.; Ball, C.; Krogstad, E. A.; Woodrow, K. A., Electrospun fibers for vaginal anti-HIV drug delivery. Antiviral Res. 2013, 100, S9-S16.
- Blakney, A. K.; Jiang, Y.; Woodrow, K. A., Application of electrospun fibers for female reproductive health. Drug Deliv. Transl. Res. 2017, 7 (6), 796-804.
- Liu, M.; Zhang, Y.; Sun, S.; Khan, A. R.; Ji, J.; Yang, M.; Zhai, G., Recent advances in electrospun for drug delivery purpose. J. Drug Target. 2019, 27 (3), 270-282.
- Wen, P.; Zong, M.-H.; Linhardt, R. J.; Feng, K.; Wu, H., Electrospinning: A novel nano-encapsulation approach for bioactive compounds. Trends Food Sci Technol. 2017, 70, 56-68.
- Katouzian, I.; Jafari, S. M., Nano-encapsulation as a promising approach for targeted delivery and controlled release of vitamins. Trends Food Sci Technol. 2016, 53, 34-48.
- Ye, C.; Chi, H., A review of recent progress in drug and protein encapsulation: Approaches, applications and challenges. Mater. Sci. Eng. C 2018, 83, 233-246.
- Paulo, F.; Santos, L., Design of experiments for microencapsulation applications: A review. Mater. Sci. Eng. C 2017, 77, 1327-1340.
- Katouzian, I.; Jafari, S. M., Nano-encapsulation as a promising approach for targeted delivery and controlled release of vitamins. Trends Food Sci Technol 2016, 53, 34-48.
- Otto, D. P.; Otto, A.; De Villiers, M. M., Differences in physicochemical properties to consider in the design, evaluation and choice between microparticles and nanoparticles for drug delivery. Expert Opin. Drug Deliv. 2015, 12 (5), 763-777.
- Martín-Villena, M.; Fernández-Campos, F.; Calpena-Campmany, A.; Bozal-de Febrer, N.; Ruiz-Martínez, M.; Clares-Naveros, B., Novel microparticulate systems for the vaginal delivery of nystatin: development and characterization. Carbohydr. Polym. 2013, 94 (1), 1-11.
- Moreno, M. A.; Gómez-Mascaraque, L. G.; Arias, M.; Zampini, I. C.; Sayago, J. E.; Ramos, L. L. P.; Schmeda-Hirschmann, G.; López-Rubio, A.; Isla, M. I., Electrosprayed chitosan microcapsules as delivery vehicles for vaginal phytoformulations. Carbohydr. Polym. 2018, 201, 425-437.
- Laelorspoen, N.; Wongsasulak, S.; Yoovidhya, T.; Devahastin, S., Microencapsulation of Lactobacillus acidophilus in zein–alginate core–shell microcapsules via electrospraying. J. Funct. Foods 2014, 7, 342-349.
- El-Hammadi, M. M.; Arias, J. L., Nanomedicine for vaginal drug delivery. In Theory and Applications of Nonparenteral Nanomedicines, Elsevier: Amsterdam, The Netherlands, 2021; pp 235-257.
- El-Hammadi, M. M.; Arias, J. L., Nanotechnology for vaginal drug delivery and targeting. In Nanoengineered Biomaterials for Advanced Drug Delivery, Elsevier: Amsterdam, The Netherlands, 2020; pp 647-682.
- Sharma, P.; Garg, S., Pure drug and polymer based nanotechnologies for the improved solubility, stability, bioavailability and targeting of anti-HIV drugs. Adv. Drug Deliv. Rev. 2010, 62 (4), 491-502.
- Joye, I. J.; McClements, D. J., Biopolymer-based nanoparticles and microparticles: Fabrication, characterization, and application. Curr Opin Colloid Interface Sci . 2014, 19 (5), 417-427.
- Leyva-Gómez, G.; Piñón-Segundo, E.; Mendoza-Muñoz, N.; Zambrano-Zaragoza, M. L.; Mendoza-Elvira, S.; Quintanar-Guerrero, D., Approaches in polymeric nanoparticles for vaginal drug delivery: a review of the state of the art. Int. J. Mol. Sci. 2018, 19 (6), 1549.
- Netsomboon, K.; Bernkop-Schnürch, A., Mucoadhesive vs. mucopenetrating particulate drug delivery. Eur. J. Pharm. Biopharm. 2016, 98, 76-89.
- Yang, M.; Yu, T.; Wang, Y. Y.; Lai, S. K.; Zeng, Q.; Miao, B.; Tang, B. C.; Simons, B. W.; Ensign, L. M.; Liu, G., Vaginal Delivery of Paclitaxel via Nanoparticles with Non‐Mucoadhesive Surfaces Suppresses Cervical Tumor Growth. Adv. Healthc. Mater. 2014, 3 (7), 1044-1052.
- Jeong, B.; Kim, S. W.; Bae, Y. H., Thermosensitive sol–gel reversible hydrogels. Adv. Drug Deliv. Rev. 2012, 64, 154-162.
- Agrawal, M.; Saraf, S.; Saraf, S.; Dubey, S. K.; Puri, A.; Gupta, U.; Kesharwani, P.; Ravichandiran, V.; Kumar, P.; Naidu, V. G. M.; Murty, U. S.; Ajazuddin; Alexander, A., Stimuli-responsive In situ gelling system for nose-to-brain drug delivery. J. Control. Release 2020, 327, 235-265.
- Bazban-Shotorbani, S.; Hasani-Sadrabadi, M. M.; Karkhaneh, A.; Serpooshan, V.; Jacob, K. I.; Moshaverinia, A.; Mahmoudi, M., Revisiting structure-property relationship of pH-responsive polymers for drug delivery applications. J. Control. Release 2017, 253, 46-63.
- Bahram, M.; Mohseni, N.; Moghtader, M., An introduction to hydrogels and some recent applications. In Emerging concepts in analysis and applications of hydrogels, IntechOpen: London, UK, 2016.
- Ruel-Gariepy, E.; Leroux, J.-C., In situ-forming hydrogels—review of temperature-sensitive systems. Eur. J. Pharm. Biopharm. 2004, 58 (2), 409-426.
- da Silva, J. B.; Cook, M. T.; Bruschi, M. L., Thermoresponsive systems composed of poloxamer 407 and HPMC or NaCMC: mechanical, rheological and sol-gel transition analysis. Carbohydr. Polym. 2020, 240, 116268.
- Sarwal, A.; Singh, G.; Singh, S.; Singh, K.; Sinha, V., Novel and effectual delivery of an antifungal agent for the treatment of persistent vulvovaginal candidiasis. J Pharm Investig. 2019, 49 (1), 135-147.
- Enggi, C. K.; Isa, H. T.; Sulistiawati, S.; Ardika, K. A. R.; Wijaya, S.; Asri, R. M.; Mardikasari, S. A.; Donnelly, R. F.; Permana, A. D., Development of thermosensitive and mucoadhesive gels of cabotegravir for enhanced permeation and retention profiles in vaginal tissue: A proof of concept study. Int. J. Pharm. 2021, 609, 121182.
- Choi, S. G.; Lee, S.-E.; Kang, B.-S.; Ng, C. L.; Davaa, E.; Park, J.-S., Thermosensitive and mucoadhesive sol-gel composites of paclitaxel/dimethyl-β-cyclodextrin for buccal delivery. PLoS One 2014, 9 (10), e109090.
- Araujo, V. H. S.; de Souza, M. P. C.; Carvalho, G. C.; Duarte, J. L.; Chorilli, M., Chitosan-based systems aimed at local application for vaginal infections. Carbohydr. Polym. 2021, 261, 117919.
- Zierden, H. C.; Josyula, A.; Shapiro, R. L.; Hsueh, H. T.; Hanes, J.; Ensign, L. M., Avoiding a Sticky Situation: Bypassing the Mucus Barrier for Improved Local Drug Delivery. Trends Mol. Med. 2021, 27, 436–450.
- Lacey, C.; Woodhall, S.; Qi, Z.; Sawant, S.; Cowen, M.; McCormack, S.; Jiang, S., Unacceptable side-effects associated with a hyperosmolar vaginal microbicide in a phase 1 trial. Int. J. STD AIDS 2010, 21 (10), 714-717.
- Liu, C.; Jiang, X.; Gan, Y.; Yu, M., Engineering nanoparticles to overcome the mucus barrier for drug delivery: Design, evaluation and state-of-the-art. Medicine in Drug Discovery 2021, 12, 100110.
- Tuğcu-Demiröz, F.; Saar, S.; Kara, A. A.; Yıldız, A.; Tunçel, E.; Acartürk, F., Development and characterization of chitosan nanoparticles loaded nanofiber hybrid system for vaginal controlled release of benzydamine. Eur. J. Pharm. Sci. 2021, 161, 105801.
- Halder, J.; Pradhan, D.; Kar, B.; Ghosh, G.; Rath, G., Nanotherapeutics approaches to overcome P-glycoprotein-mediated multi-drug resistance in cancer. Nanomed.: Nanotechnol. Biol. Med. 2021, 40, 102494.
- Kim, Y.-T.; Shin, B.-K.; Garripelli, V. K.; Kim, J.-K.; Davaa, E.; Jo, S.; Park, J.-S., A thermosensitive vaginal gel formulation with HPγCD for the pH-dependent release and solubilization of amphotericin B. Eur. J. Pharm. Sci. 2010, 41 (2), 399-406.
- Chindamo, G.; Sapino, S.; Peira, E.; Chirio, D.; Gallarate, M., Recent Advances in Nanosystems and Strategies for Vaginal Delivery of Antimicrobials. Nanomaterials 2021, 11 (2), 311.
- Ramyadevi, D.; Rajan, K. S.; Vedhahari, B. N.; Ruckmani, K.; Subramanian, N., Heterogeneous polymer composite nanoparticles loaded in situ gel for controlled release intra-vaginal therapy of genital herpes. Colloids Surf. B 2016, 146, 260-270.
- Iqbal, R.; Qureshi, O. S.; Yousaf, A. M.; Raza, S. A.; Sarwar, H. S.; Shahnaz, G.; Saleem, U.; Sohail, M. F., Enhanced solubility and biopharmaceutical performance of atorvastatin and metformin via electrospun polyvinylpyrrolidone-hyaluronic acid composite nanoparticles. Eur. J. Pharm. Sci. 2021, 161, 105817.
- Deshkar, S. S.; Palve, V. K., Formulation and development of thermosensitive cyclodextrin-based in situ gel of voriconazole for vaginal delivery. J Drug Deliv Sci Technol. 2019, 49, 277-285.
- Shaaban, O. M.; Fetih, G. N.; Abdellah, N. H.; Ismail, S.; Ibrahim, M. A.; Ibrahim, E. s. A., Pilot randomized trial for treatment of bacterial vaginosis using in situ forming metronidazole vaginal gel. J. Obstet. Gynaecol. Res. 2011, 37 (7), 874-881.
- Shi, S.; Nguyen, P. K.; Cabral, H. J.; Diez-Barroso, R.; Derry, P. J.; Kanahara, S. M.; Kumar, V. A., Development of peptide inhibitors of HIV transmission. Bioact. Mater. 2016, 1 (2), 109-121.
- Shaikh, R. P.; Pillay, V.; Choonara, Y. E.; du Toit, L. C.; Ndesendo, V. M.; Bawa, P.; Cooppan, S., A review of multi-responsive membranous systems for rate-modulated drug delivery. AAPS PharmSciTech 2010, 11 (1), 441-459.
- Navath, R. S.; Menjoge, A. R.; Dai, H.; Romero, R.; Kannan, S.; Kannan, R. M., Injectable PAMAM dendrimer–PEG hydrogels for the treatment of genital infections: formulation and in vitro and in vivo evaluation. Mol. Pharm. 2011, 8 (4), 1209-1223.
- Nie, L.; Zou, P.; Dong, J.; Sun, M.; Ding, P.; Han, Y.; Ji, C.; Zhou, Q.; Yuan, H.; Suo, J., Injectable vaginal hydrogels as a multi-drug carrier for contraception. Appl. Sci. 2019, 9 (8), 1638.
- Liu, Y.; Yang, F.; Feng, L.; Yang, L.; Chen, L.; Wei, G.; Lu, W., In vivo retention of poloxamer-based in situ hydrogels for vaginal application in mouse and rat models. Acta Pharm. Sin. B. 2017, 7 (4), 502-509.
- das Neves, J.; Nunes, R.; Rodrigues, F.; Sarmento, B., Nanomedicine in the development of anti-HIV microbicides. Adv. Drug Deliv. Rev. 2016, 103, 57-75.
- Lakshmi, Y. S.; Kumar, P.; Kishore, G.; Bhaskar, C.; Kondapi, A. K., Triple combination MPT vaginal microbicide using curcumin and efavirenz loaded lactoferrin nanoparticles. Sci. Rep. 2016, 6 (1), 1-13.
- Mesquita, L.; Galante, J.; Nunes, R.; Sarmento, B.; das Neves, J., Pharmaceutical vehicles for vaginal and rectal administration of anti-HIV microbicide nanosystems. Pharmaceutics 2019, 11 (3), 145.
- Li, Z.; Cho, S.; Kwon, I. C.; Janát-Amsbury, M. M.; Huh, K. M., Preparation and characterization of glycol chitin as a new thermogelling polymer for biomedical applications. Carbohydr. Polym. 2013, 92 (2), 2267-2275.
- Almomen, A.; Cho, S.; Yang, C.-H.; Li, Z.; Jarboe, E. A.; Peterson, C. M.; Huh, K. M.; Janát-Amsbury, M. M., Thermosensitive progesterone hydrogel: a safe and effective new formulation for vaginal application. Pharm. Res. 2015, 32 (7), 2266-2279.
- Arun Karthick, R.; Ramya Devi, D.; Vedha Hari, B. N., Investigation of sustained release mucoadhesive in-situ gel system of Secnidazole for the persistent treatment of vaginal infections. J Drug Deliv Sci Technol. 2018, 43, 362-368.
- Patel, P.; Patel, P., Formulation and evaluation of clindamycin HCL in situ gel for vaginal application. Int. J. Pharm. Investig. 2015, 5 (1), 50.
- Vandenhaute, M.; Schelfhout, J.; Van Vlierberghe, S.; Mendes, E.; Dubruel, P., Cross-linkable, thermo-responsive Pluronic® building blocks for biomedical applications: Synthesis and physico-chemical evaluation. Eur. Polym. J. 2014, 53, 126-138.
- Lu, C.; Liu, M.; Fu, H.; Zhang, W.; Peng, G.; Zhang, Y.; Cao, H.; Luo, L., Novel thermosensitive in situ gel based on poloxamer for uterus delivery. Eur. J. Pharm. Sci. 2015, 77, 24-28.
- Soliman, G. M.; Fetih, G.; Abbas, A. M., Thermosensitive bioadhesive gels for the vaginal delivery of sildenafil citrate: in vitro characterization and clinical evaluation in women using clomiphene citrate for induction of ovulation. Drug Dev. Ind. Pharm. 2017, 43 (3), 399-408.
- Abd Ellah, N. H.; Abouelmagd, S. A.; Abbas, A. M.; Shaaban, O. M.; Hassanein, K. M. A., Dual-responsive lidocaine in situ gel reduces pain of intrauterine device insertion. Int. J. Pharm. 2018, 538 (1), 279-286.
- Yang, T.-T.; Cheng, Y.-Z.; Qin, M.; Wang, Y.-H.; Yu, H.-L.; Wang, A.-L.; Zhang, W.-F., Thermosensitive chitosan hydrogels containing polymeric microspheres for vaginal drug delivery. Biomed Res. Int. 2017, 3564060.
- Hu, X.; Tang, Y.; Wang, Q.; Li, Y.; Yang, J.; Du, Y.; Kennedy, J. F., Rheological behaviour of chitin in NaOH/urea aqueous solution. Carbohydr. Polym. 2011, 83 (3), 1128-1133.
- Dos Santos, A. M.; Carvalho, S. G.; Araujo, V. H. S.; Carvalho, G. C.; Gremião, M. P. D.; Chorilli, M., Recent advances in hydrogels as strategy for drug delivery intended to vaginal infections. Int. J. Pharm. 2020, 590, 119867.
- Akash, M. S. H.; Rehman, K., Recent progress in biomedical applications of Pluronic (PF127): Pharmaceutical perspectives. J. Control. Release 2015, 209, 120-138.
- Echeverria, C.; Fernandes, S. N.; Godinho, M. H.; Borges, J. P.; Soares, P. I., Functional stimuli-responsive gels: Hydrogels and microgels. Gels 2018, 4 (2), 54.
- Pandey, P.; Cabot, P. J.; Wallwork, B.; Panizza, B. J.; Parekh, H. S., Formulation, functional evaluation and ex vivo performance of thermoresponsive soluble gels-A platform for therapeutic delivery to mucosal sinus tissue. Eur. J. Pharm. Sci. 2017, 96, 499-507.
- Russo, E.; Villa, C., Poloxamer hydrogels for biomedical applications. Pharmaceutics 2019, 11 (12), 671.
- Ci, L.; Huang, Z.; Liu, Y.; Liu, Z.; Wei, G.; Lu, W., Amino-functionalized poloxamer 407 with both mucoadhesive and thermosensitive properties: preparation, characterization and application in a vaginal drug delivery system. Acta Pharm. Sin. B. 2017, 7 (5), 593-602.
- Barros, S. C.; da Silva, A. A.; Costa, D. B.; Cesarino, I.; Costa, C. M.; Lanceros-Méndez, S.; Pawlicka, A.; Silva, M. M., Thermo-sensitive chitosan–cellulose derivative hydrogels: swelling behaviour and morphologic studies. Cellulose. 2014, 21 (6), 4531-4544.
- Klouda, L., Thermoresponsive hydrogels in biomedical applications: A seven-year update. Eur. J. Pharm. Biopharm. 2015, 97, 338-349.
- Chatterjee, S.; Hui, P. C.-l., Review of Applications and Future Prospects of Stimuli-Responsive Hydrogel Based on Thermo-Responsive Biopolymers in Drug Delivery Systems. Polymers 2021, 13 (13), 2086.
- Chai, Q.; Jiao, Y.; Yu, X., Hydrogels for biomedical applications: their characteristics and the mechanisms behind them. Gels 2017, 3 (1), 6.
- Chen, D.; Yu, H.; Sun, K.; Liu, W.; Wang, H., Dual thermoresponsive and pH-responsive self-assembled micellar nanogel for anticancer drug delivery. Drug Deliv. 2014, 21 (4), 258-64.
- Qin, C.; Zhou, J.; Zhang, Z.; Chen, W.; Hu, Q.; Wang, Y., Convenient one-step approach based on stimuli-responsive sol-gel transition properties to directly build chitosan-alginate core-shell beads. Food Hydrocoll. 2019, 87, 253-259.
- Cirri, M.; Maestrelli, F.; Scuota, S.; Bazzucchi, V.; Mura, P., Development and microbiological evaluation of chitosan and chitosan-alginate microspheres for vaginal administration of metronidazole. Int. J. Pharm. 2021, 598, 120375.
- Rodriguez-Tenreiro, C.; Diez-Bueno, L.; Concheiro, A.; Torres-Labandeira, J. J.; Alvarez-Lorenzo, C., Cyclodextrin/carbopol micro-scale interpenetrating networks (ms-IPNs) for drug delivery. J. Control. Release 2007, 123 (1), 56-66.
- Migliozzi, S.; Angeli, P.; Mazzei, L., Gelation kinetics of non-aqueous Carbopol dispersions. Colloids Surf. B 2019, 577, 84-95.
- Singh, V. K.; Anis, A.; Banerjee, I.; Pramanik, K.; Bhattacharya, M. K.; Pal, K., Preparation and characterization of novel carbopol based bigels for topical delivery of metronidazole for the treatment of bacterial vaginosis. Mater. Sci. Eng. C 2014, 44, 151-158.
- Ching, S. H.; Bansal, N.; Bhandari, B., Alginate gel particles–A review of production techniques and physical properties. Crit. Rev. Food Sci. Nutr. 2017, 57 (6), 1133-1152.
- Cao, L.; Lu, W.; Mata, A.; Nishinari, K.; Fang, Y., Egg-box model-based gelation of alginate and pectin: A review. Carbohydr. Polym. 2020, 242, 116389.
- Mishra, R.; Soni, K.; Mehta, T., Mucoadhesive vaginal film of fluconazole using cross-linked chitosan and pectin. J. Therm. Anal. Calorim. 2017, 130 (3), 1683-1695.
- Bakke, A. J.; Zaveri, T.; Higgins, M. J.; Ziegler, G. R.; Hayes, J. E., Design aspects of vaginal applicators that influence acceptance among target users. Sci. Rep. 2021, 11 (1), 1-11.
- Montesino, M.; Labrie, F.; Archer, D. F.; Zerhouni, J.; Côté, I.; Lavoie, L.; Beauregard, A.; Martel, C.; Vaillancourt, M.; Moyneur, E., Evaluation of the acceptability of intravaginal prasterone ovule administration using an applicator. Gynecol. Endocrinol. 2016, 32 (3), 240-245.
- Omar, R. F.; Trottier, S.; Brousseau, G.; Ouellet, C.; Danylo, A.; Ong, T.; Bergeron, M. G., Universal vaginal applicator for the uniform distribution of vaginal gel and cream formulations: a magnetic resonance imaging study. J Obstet Gynaecol Can. 2014, 36 (1), 42-50.
- Brache, V.; Cohen, J. A.; Cochon, L.; Alvarez, F., Evaluating the clinical safety of three vaginal applicators: a pilot study conducted in the Dominican Republic. Contraception 2006, 73 (1), 72-77.
- Brunner, H.; Theodor, R. A., Multiple use applicator for vaginal tablets/vaginal inserts: compliance verification and suitability studies. BMC Womens Health 2020, 20 (1), 1-9.
This entry is adapted from the peer-reviewed paper 10.3390/gels8020099