Chronic lymphocytic leukemia (CLL) is an indolent form of B-cell lymphoid malignancy that predominantly affects older individuals. Bruton’s tyrosine kinase (BTK) is a member of the TEC kinase family. It plays an important role in treatment of malignant B lymphocyte proliferation and improvement of survival of patients with CLL.
- CLL
- BTK
- BTK inhibitors
- ibrutinib
- acalabrutinib
- zanubrutinib
- spebrutinib
- tirabrutinib
- orelabrutinib
- TG-1701
- DTRMWXHS-12
- pirtobrutinib
- vecabrutynib
- fenebrutinib
- nemtabrutinib
1. Introduction
|
BTKi |
Binding |
T1/2 [hours] |
IC50 [nM] |
Dosing |
Clinical Trials in CLL |
|---|---|---|---|---|---|
|
Ibrutinib (PCYC-1102) |
Covalent irreversible C481 |
4–8 |
0.5 |
420 mg |
NCT04771507 NCT03513562 NCT02912754 |
|
Acalabrutinib (ACP-196) |
Covalent irreversible C481 |
0.9 |
5.1 |
100 mg twice a day |
NCT04008706 NCT04930536 NCT04722172 |
|
Zanubrutinib (BGB-3111) |
Covalent irreversible C481 |
2–4 |
0.5 |
160 or 320 mg twice a day |
NCT04116437 NCT04458610 NCT03824483 NCT04282018 NCT04515238 NCT03336333 |
|
Spebrutinib (CC-292) |
Covalent irreversible C481 |
8–24 |
<0.5 |
1000 mg |
NCT02031419 |
|
Tirabrutinib (ONO/GS-4059) |
Covalent irreversible C481 |
NA |
5.6 |
80 mg |
NCT03740529 NCT03162536 |
|
Orelabrutinib (ICP-022) |
Covalent irreversible C481 |
~1.5–4 h |
1.6 |
150 mg |
NCT03493217 NCT04014205 |
|
SHR1459 (TG-1701) |
Covalent irreversible C481 |
NA |
3 |
300 mg |
NCT03671590; NCT04806035 |
|
DTRMWXHS-12 (DTRM-12) |
Covalent irreversible C481 |
~4 |
NA |
200 mg |
NCT02900716 NCT04305444 |
|
Pirtobrutinib (LOXO-305) |
Non-covalent reversible |
NA |
0.85 |
200 mg |
NCT05023980 NCT04965493 NCT05024045 NCT04666038 |
|
Vecabrutinib (SNS-062) |
Non-covalent reversible |
6.6-8 |
24 |
25 mg escalated to 500 mg |
NCT03037645 |
|
Fenebrutinib (GDC-0853) |
Non-covalent reversible |
2.2 |
0.91 |
200 mg twice a day |
NCT01991184 |
|
Nemta brutinib (ARQ 531) |
Non-covalent reversible |
NA |
0.85 |
65-100 mg |
NCT04728893 NCT03162536 |
Abbreviations: BTKi—Bruton tyrosine kinase inhibitor, NA—not available.
2. Mechanism of Action
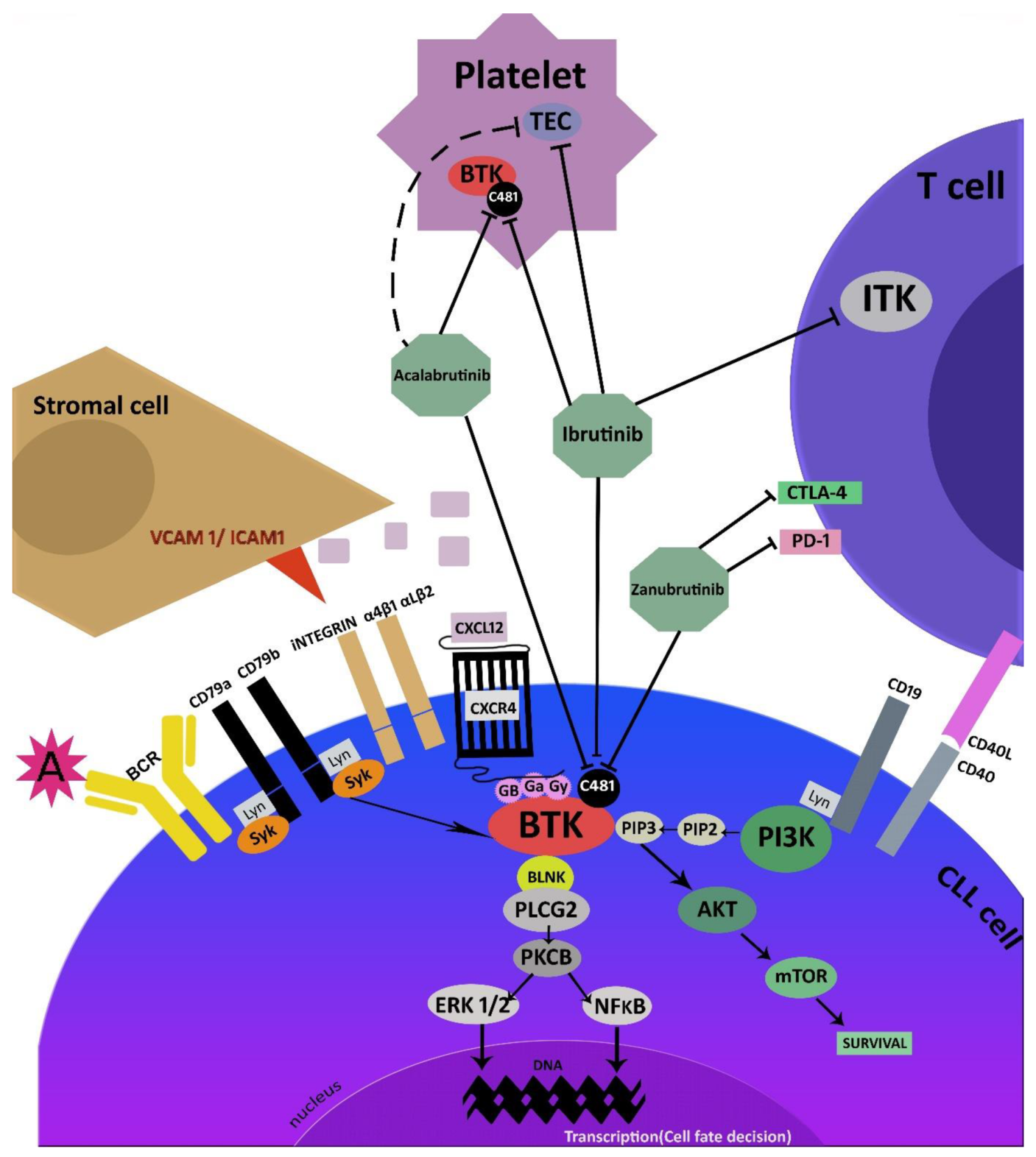
3. Irreversible Covalent BTK Inhibitors
3.1. Ibrutnib
3.2. Acalabrutinib
3.3. Zanubrutinib
3.4. Other Irreversible BTK Inhibitors
3.4.1. Spebrutinib
3.4.2. Orelabrutinib
3.4.3. Tirabrutinib
3.4.4. SHR1459
3.4.5. DTRMWXHS-12
4. Reversible BTK Inhibitors
4.1. Pirtobrutinib
4.2. Vecabrutinib
4.3. Fenebrutinib
4.4. Nemtabrutinib
5. Resistance to BTK Inhibitors
6. Adverse Events
6.1. Bleeding and Bruising
6.2. Cardiovascular Complications
6.3. Cytopenias
6.4. Infections
6.5. Arthralgias and Myalgias
6.6. Dermatologic Complications
6.7. Headaches
6.8. Diarrhea
This entry is adapted from the peer-reviewed paper 10.3390/cancers14030771
References
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Dohner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. iwCLL guidelines for diagnosis, indications for treatment, response assessment, and supportive man-agement of CLL. Blood 2018, 131, 2745–2760.
- Eichhorst, B.; Robak, T.; Montserrat, E.; Ghia, P.; Niemann, C.; Kater, A.; Gregor, M.; Cymbalista, F.; Buske, C.; Hillmen, P.; et al. Chronic lymphocytic leukaemia: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2020, 32, 23–33.
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Brest, A.; Yu, M.; Ruhl, J.; Tatalovich, Z.; Mariotto, A.; Lewis, D.R.; et al. SEER Cancer Statistics Review, 1975–2016; National Cancer Institute: Bethesda, MD, USA, 2019. Available online: https://seer.cancer.gov/csr/1975_2016/ (accessed on 9 April 2020).
- Alrawashdh, N.; Sweasy, J.; Erstad, B.; McBride, A.; Persky, D.O.; Abraham, I. Survival trends in chronic lymphocytic leukemia across treatment eras: US SEER database analysis (1985–2017). Ann. Hematol. 2021, 100, 2501–2512.
- Brullo, C.; Villa, C.; Tasso, B.; Russo, E.; Spallarossa, A. Btk Inhibitors: A medicinal chemistry and drug delivery perspective. Int. J. Mol. Sci. 2021, 22, 7641.
- Wen, T.; Wang, J.; Shi, Y.; Qian, H.; Liu, P. Inhibitors targeting Bruton’s tyrosine kinase in cancers: Drug development advances. Leukemia 2021, 35, 312–332.
- Robak, T.; Burger, J.A.; Tedeschi, A.; Barr, P.M.; Owen, C.; Bairey, O.; Hillmen, P.; Simpson, D.; Grosicki, S.; Devereux, S.; et al. Single-agent ibrutinib versus chemoimmunotherapy regimens for treatment-naïve patients with chronic lymphocytic leu-kemia: A cross-trial comparison of phase 3 studies. Am. J. Hematol. 2018, 93, 1402–1410.
- Moreno, C.; Greil, R.; Demirkan, F.; Tedeschi, A.; Anz, B.; Larratt, L.; Simkovic, M.; Novak, J.; Strugov, V.; Gill, D.; et al. First-line treatment of chronic lymphocytic leukemia with ibrutinib plus obinutuzumab versus chlorambucil plus obinutuzumab: Final analysis of the randomized, phase 3 iLLUMINATE trial. Haematologica 2022, 107.
- Burger, J.A.; Tedeschi, A.; Barr, P.M.; Robak, T.; Owen, C.; Ghia, P.; Bairey, O.; Hillmen, P.; Bartlett, N.L.; Li, J.; et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N. Engl. J. Med. 2015, 373, 2425–2437.
- Byrd, J.C.; Brown, J.R.; O’Brien, S.; Barrientos, J.C.; Kay, N.E.; Reddy, N.M.; Coutre, D.; Tam, C.S.; Mulligan, S.P.; Jaeger, U.; et al. Ibrutinib versus ofa-tumumab in previously treated chronic lymphoid leukemia. N. Engl. J. Med. 2014, 371, 213–223.
- Ghia, P.; Pluta, A.; Wach, M.; Lysak, D.; Kozak, T.; Simkovic, M.; Kaplan, P.; Kraychok, I.; Illes, A.; De La Serna, J.; et al. Acalabrutinib vs Rituximab plus Idelalisib (IdR) or Bendamustine (BR) by investigator choice in relapsed/refractory (RR) chronic lymphocytic leukemia: Phase 3 ASCEND study. Hematol. Oncol. 2019, 37, 86–87.
- Sharman, J.P.; Egyed, M.; Jurczak, W.; Skarbnik, A.; Pagel, J.M.; Flinn, I.W.; Kamdar, M.; Munir, T.; Walewska, R.; Corbett, G.; et al. Acalabrutinib with or without obinutuzumab versus chlorambucil and obinutuzmab for treatment-naive chronic lymphocytic leukaemia (ELEVATE TN): A randomised, controlled, phase 3 trial. Lancet 2020, 395, 1278–1291.
- Zain, R.; Vihinen, M. Structure-Function Relationships of Covalent and Non-Covalent BTK Inhibitors. Front. Immunol. 2021, 12, 694853.
- Estupiñán, H.Y.; Wang, Q.; Berglöf, A.; Schaafsma, G.C.P.; Shi, Y.; Zhou, L.; Mohammad, D.K.; Yu, L.; Vihinen, M.; Zain, R.; et al. BTK gatekeeper residue variation combined with cysteine 481 substitution causes super-resistance to irreversible inhibitors acalabrutinib, ibrutinib and zanubrutinib. Leukemia 2021, 35, 1317–1329.
- Byrd, J.; Furman, R.R.; Coutre, S.E.; Flinn, I.W.; Burger, J.A.; Blum, K.A.; Grant, B.; Sharman, J.P.; Coleman, M.; Wierda, W.G.; et al. Single-agent ibrutinib in treatment-naive and relapsed/refractory chronic lymphocytic leukemia: A 5-year experience. Blood 2018, 131, 1910–1919.
- Byrd, J.C.; Hillmen, P.; Ghia, P.; Kater, A.P.; Chanan-Khan, A.; Furman, R.R.; O’Brien, S.; Yenerel, M.N.; Illés, A.; Kay, N.; et al. Acalabrutinib versus ibrutinib in previously treated chronic lymphocytic leukemia: Results of the first randomized phase III trial. J. Clin. Oncol. 2021, 39, 3441–3452.
- Hillmen., P.; Eichhorst., B.; Brown, J.R.; Lamanna, N.; O’Brien, S.; Tam, C.S.; Qiu, L.; Kazmierczak, M.; Zhou, K.; Šimkovič, M.; et al. First Interim Analysis of Alpine Study: Results of a Phase 3 Randomized Study of Zanubrutinib vs. Ibrutinib in Patients with Re-lapsed/Refractory Chronic Lymphocytic Leukemia/Small Lymphocytic Lymphoma; EHA Library: Hague, The Netherlands, 2021; 330170; LB1900; Available online: https://library.ehaweb.org/eha/2021/eha2021-virtualcon-gress/330170/peter.hillmen.first.interim.analysis.of.alpine.study.results.of.a.phase.3.html?f=listing%3D0%2Abrowseby%3D8%2Asortby%3D1%2Asearch%3Dlb1900 (accessed on 11 June 2021).
- Ahn, I.E.; Brown, J.R. Targeting Bruton’s tyrosine kinase in CLL. Front. Immunology 2021, 12, 687458.
- Woyach, J.A.; Bojnik, E.; Ruppert, A.S.; Stefanovski, M.R.; Goettl, V.M.; Smucker, K.A.; Smith, L.L.; Dubovsky, L.S.J.; Towns, W.H.; MacMurray, M.M.; et al. Bruton’s tyrosine kinase (BTK) function is important to the development and expansion of chronic lymphocytic leukemia (CLL). Blood 2014, 123, 1207–1213.
- Gauld, S.B.; Porto, J.M.D.; Cambier, J.C. B cell antigen receptor signaling: Roles in cell development and disease. Science 2002, 296, 1641–1642.
- Davis, R.E.; Ngo, V.N.; Lenz, G.; Tolar, P.; Young, R.M.; Romesser, P.B.; Kohlhammer, H.; Lamy, L.; Zhao, H.; Yang, Y.; et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature 2010, 463, 88–92.
- Kaptein, A.; de Bruin, G.; Emmelot-van Hoek, M.; van de Kar, B.; de Jong, A.; Gulrajani, M.; Demont, D.; Covey, T.; Mittag, D.; Barf, T. Potency and selectivity of BTK inhibitors in clinical development for B-Cell malignancies. Blood 2018, 132, 1871.
- Alsadhan, A.; Cheung, J.; Gulrajani, M.; Gaglione, E.M.; Nierman, P.; Hamdy, A.; Izumi, R.; Bibikova, E.; Patel, P.; Sun, C.; et al. Pharmacodynamic Analysis of btk inhibition in patients with chronic lymphocytic leukemia treated with acalabrutinib. Clin. Cancer Res. 2020, 26, 2800–2809.
- Lynch, T.J.; Kim, E.S.; Eaby, B.; Garey, J.; West, D.P.; Lacouture, M.E.; Zhu, A.X.; Fuchs, C.S.; Clark, J.W.; Muzikansky, A.; et al. Epidermal growth factor receptor inhibitor–associated cutaneous toxicities: An evolving paradigm in clinical management. Oncologist 2007, 12, 610–621.
- Barf, T.; Covey, T.; Izumi, R.; Van De Kar, B.; Gulrajani, M.; Van Lith, B.; Van Hoek, M.; De Zwart, E.; Mittag, D.; Demont, D.; et al. Acalabrutinib (ACP-196): A covalent Bruton tyrosine kinase inhibitor with a differentiated selectivity and in vivo potency profile. J. Pharmacol. Exp. Ther. 2017, 363, 240–252.
- Pavlasova, G.; Mraz, M. The regulation and function of CD20: An “enigma” of B-cell biology and targeted therapy. Haema-tologica 2020, 105, 1494–1506.
- Cervantes-Gomez, F.; Lamothe, B.; Woyach, J.A.; Wierda, W.G.; Keating, M.J.; Balakrishnan, K.; Gandhi, V. Pharmacological and Protein Profiling Suggests Venetoclax (ABT-199) as optimal partner with ibrutinib in chronic lymphocytic leukemia. Clin. Cancer Res. 2015, 21, 3705–3715.
- Haselager, M.V.; Kielbassa, K.; Ter Burg, J.; Bax, D.J.C.; Fernandes, S.M.; Borst, J.; Tam, C.; Forconi, F.; Chiodin, G.; Brown, J.R.; et al. Changes in Bcl-2 members after ibrutinib or venetoclax uncover functional hierarchy in determining resistance to venetoclax in CLL. Blood 2020, 136, 2918–2926.
- Pan, Z.; Scheerens, H.; Li, S.-J.; Schultz, B.E.; Sprengeler, P.A.; Burrill, L.C.; Mendonca, R.V.; Sweeney, M.D.; Scott, K.C.K.; Grothaus, P.G.; et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2006, 2, 58–61.
- Honigberg, L.A.; Smith, A.M.; Sirisawad, M.; Verner, E.; Loury, D.; Chang, B.; Li, S.; Pan, Z.; Thamm, D.H.; Miller, R.A.; et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proc. Natl. Acad. Sci. USA 2010, 107, 13075–13080.
- Brown, J.R. How I treat CLL patients with ibrutinib. Blood 2018, 131, 379–386.
- Tam, C.; Giannopoulos, K.; Jurczak, W.; Šimkovič, M.; Shadman, M.; Österborg, A.; Laurenti, L.; Walker, P.; Opat, S.; Chan, H.; et al. SEQUOIA: Results of a phase 3 randomized study of zanubrutinib versus bendamustine + rituximab (BR) in patients with treatment-naïve (TN) chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL). Blood 2021, 138, 396.
- Robak, T.; Robak, P. BCR signaling in chronic lymphocytic leukemia and related inhibitors currently in clinical studies. Int. Rev. Immunol. 2013, 32, 358–376.
- Tam, C.; Grigg, A.P.; Opat, S.; Ku, M.; Gilbertson, M.; Anderson, M.A.; Seymur, J.F.; Ritchie, D.S.; Dircoletto, C.; Dimowski, B.; et al. The BTK inhibitor, BGB-3111, is safe, tolerable, and highly active in patients with relapsed/refractory B-cell malignancies: Initial report of a phase 1 first-in-human trial. Blood 2015, 126, 832.
- Flinsenberg, T.W.; Tromedjo, C.C.; Hu, N.; Liu, Y.; Guo, Y.; Thia, K.Y.; Noori, T.; Song, X.; Yeang, H.X.A.; Tantalo, D.G.; et al. Differential effects of BTK inhibitors ibrutinib and zanubrutinib on NK-cell effector function in patients with mantle cell lymphoma. Haematologica 2019, 105, e76–e79.
- Zou, Y.; Zhu, H.; Li, X.; Xia, Y.; Miao, K.; Zhao, S.; Wu, Y.; Wang, L.; Xu, W.; Li, J. The impacts of zanubrutinib on immune cells in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma. Hematol. Oncol. 2019, 37, 392–400.
- Chen, W.; Loury, D.J.; Mody, T.D. Preparation of N- Amides as Inhibitors of Bruton’s Tyrosine Kinase. U.S. Patent WO 2013173518 A1, 21 November 2013.
- Schafer, P.H.; Kivitz, A.J.; Ma, J.; Korish, S.; Sutherland, D.; Li, L.; Azaryan, A.; Kosek, J.; Adams, M.; Capone, L.; et al. Spebrutinib (CC-292) affects markers of B cell activation, chemotaxis, and osteoclasts in patients with rheumatoid arthritis: Results from a mechanistic study. Rheumatol. Ther. 2019, 7, 101–119.
- Evans, E.K.; Tester, R.; Aslanian, S.; Karp, R.; Sheets, M.; Labenski, M.T.; Witowski, S.R.; Lounsbury, H.; Chaturvedi,, P.; Mazdiyasni,, H.; et al. Inhibition of Btk with CC-292provides early pharmacodynamic assessment ofactivity in mice and humans. J. Pharmacoll. Exp. Ther. 2013, 346, 219–228.
- Brown, J.R.; Harb, W.A.; Hill, B.T.; Gabrilove, J.; Sharman, J.P.; Schreeder, M.T.; Barr, P.M.; Foran, J.M.; Miller, T.P.; Burger, J.A.; et al. Phase I study of single-agent CC-292, a highly selective Brutons tyrosine kinase inhibitor, in relapsed/refractory chronic lymphocytic leukemia. Haematologica 2016, 101, e295–e298.
- Zhang, B.; Zhao, R.; Liang, R.; Gao, Y.; Liu, R.; Chen, X.; Lu, Z.; Wang, Z.; Yu, L.; Shakib, S.; et al. Abstract CT132: Orelabrutinib, a potent and selective Bruton’s tyrosine kinase inhibitor with superior safety profile and excellent PK/PD properties. Tumor Biol. 2020, 80, CT132.
- Xu, W.; Song, Y.; Li, Z.; Yang, S.; Liu, L.; Hu, Y.; Zhang, W.; Zhou, J.; Gao, S.; Ding, K.; et al. Safety, Tolerability and efficacy of orelabrutinib, once a day, to treat chinese patients with relapsed or refractory chronic lymphocytic leukemia/small cell leukemia. Blood 2019, 134, 4319.
- Dhillon, S. Orelabrutinib: First Approval. Drugs 2021, 81, 503–507.
- Walter, H.S.; Rule, S.A.; Dyer, M.J.S.; Karlin, L.; Jones, C.; Cazin, B.; Quittet, P.; Shah, N.; Hutchinson, C.V.; Honda, H.; et al. A phase 1 clinical trial of the selective BTK inhibitor ONO/GS-4059 in relapsed and refractory mature B-cell malignancies. Blood 2016, 127, 411–419.
- Liu, Y.; Yao, Q.; Zhang, F. Diagnosis, prognosis and treatment of primary central nervous system lymphoma in the elderly population (Review). Int. J. Oncol. 2021, 58, 371–387.
- Walter, H.S.; Jayne, S.; Rule, S.A.; Cartron, G.; Morschhauser, F.; Macip, S.; Karlin, L.; Jones, C.; Herbaux, C.; Quittet, P.; et al. Long-term follow-up of patients with CLL treated with the selective Bruton’s tyrosine kinase inhibitor ONO/GS-4059. Blood 2017, 129, 2808–2810.
- Ribeiro, M.L.; Reyes-Garau, D.; Vinyoles, M.; Profitós Pelejà, N.; Santos, J.; Armengol, M.; Fernández-Serrano, M.; Sedó Mor, A.; Bech-Serra, J.J.; Blecua, P.; et al. Antitumor activity of the novel BTK inhibitor TG-1701 is associated with disruption of Ikaros signaling in patients with B-cell Non-Hodgkin lymphoma. Clin. Cancer. Res. 2021, 27, 6591–6601.
- Cheah, C.Y.; Wickham, N.; Jurczak, W.; Lasica, M.; Wróbel, T.; Walewski, J.; Yannakou, C.K.; Lewis, K.L.; Dlugosz-Danecka, M.; Giannopoulos, K.; et al. Clinical activity of TG-1701, as monotherapy and in combination with ublituximab and umbralisib (U2), in patients with B-cell malignancies. Blood 2020, 136, 1130.
- Gill, J.; He, W.; Schuster, S.J.; Brander, D.M.; Chatburn, E.; Kennard, K.; Anderson, B.D.; Nasta, S.; Landsburg, D.J.; Porter, D.L.; et al. A phase Ia/Ib study of a novel BTK inhibitor, DTRMWXHS-12 (DTRM-12), and combination products, with everolimus and pomalidomide, in pts with CLL or other B-cell lymphomas. J. Clin. Oncol. 2017, 35, TPS7570.
- Mato, A.R.; Schuster, S.J.; Foss, F.M.; Isufi, I.; Ding, W.; Brander, D.M.; Sitlinger, A.; Tun, H.W.; Moustafa, M.A.; Kennard, K.; et al. A Phase Ia/Ib study exploring the synthetic lethality of the orally administered novel BTK inhibitor, Dtrmwxhs-12 (DTRM-12), in combination with everolimus and pomalidomide in patients with relapsed/refractory CLL, DLBCL or other B-cell lymphomas. Blood 2019, 134, 810.
- Tambaro, F.P.; De Novellis, D.; Wierda, W.G. The Role of BTK Inhibition in the Treatment of Chronic Lymphocytic Leukemia: A Clinical View. J. Exp. Pharmacol. 2021, 13, 923–935.
- Lewis, K.L.; Cheah, C.Y.J. Non-covalent BTK inhibitors-the new BTKids on the block for B-cell malignancies. Pers. Med. 2021, 11, 764.
- Michot, J.-M.; Ribrag, V. Pirtobrutinib shows evidence to inaugurate a third generation of BTK inhibitors. Lancet 2021, 397, 855–857.
- Mato, A.R.; Shah, N.N.; Jurczak, W.; Cheah, C.Y.; Pagel, J.M.; Woyach, J.A.; Fakhri, B.; Eyre, T.A.; Lamanna, N.; Patel, M.R.; et al. Pirtobrutinib in relapsed or refractory B-cell malignancies (BRUIN): A phase 1/2 study. Lancet 2021, 397, 892–901.
- Binnerts, M.E.; Otipoby, K.L.; Hopkins, B.T. SNS-062 is a potent noncovalent BTK inhibitor with comparable activity against wide type BTK and BTK with an acquired resistance mutation. Mol. Cancer Ther. 2015, 14, C186.
- Burger, J.A. Bruton tyrosine kinase inhibitors: Present and future. Cancer J. 2019, 25, 386–393.
- Crawford, J.J.; Johnson, A.R.; Misner, D.L.; Belmont, L.D.; Castanedo, G.; Choy, R.; Coraggio, M.; Dong, L.; Eigenbrot, C.; Erickson, R.; et al. Discovery of GDC-0853: A potent, selective, and noncovalent bruton’s tyrosine kinase inhibitor in early clinical development. J. Med. Chem. 2018, 61, 2227–2245.
- Reiff, S.D.; Muhowski, E.M.; Guinn, D.; Lehman, A.; Fabian, C.A.; Cheney, C.; Mantel, R.; Smith, L.; Johnson, A.J.; Young, W.B.; et al. Noncovalent inhibition of C481S Bruton tyrosine kinase by GDC-0853: A new treatment strategy for ibrutinib-resistant CLL. Blood 2018, 132, 1039–1049.
- Cohen, S.; Tuckwell, K.; Katsumoto, T.R.; Zhao, R.; Galanter, J.; Lee, C.K.; Rae, J.; Toth, B.; Ramamoorthi, N.; Hackney, J.A.; et al. Fenebrutinib versus placebo or adalimumab in rheumatoid arthritis: A randomized, double-blind, phase II trial (ANDES Study). Arthritis Rheumatol. 2020, 72, 1435–1446.
- Byrd, J.C.; Smith, S.; Wagner-Johnston, N.; Sharman, J.; Chen, A.I.; Advani, R.; Augustson, B.; Marlton, P.; Commerford, S.R.; Okrah, K.; et al. First-in-human phase 1 study of the BTK inhibitor GDC-0853 in relapsed or refractory B-cell NHL and CLL. Oncotarget 2018, 9, 13023–13035.
- Shirley, M. Bruton tyrosine kinase inhibitors in B-cell malignancies: Their use and differential features. Target Oncol. 2022, 17, 69–84.
- Reiff, S.D.; Mantel, R.; Smith, L.L.; Greene, J.; Muhowski, E.M.; Fabian, C.A.; Goettl, V.M.; Tran, M.; Harrington, B.; Rogers, K.A.; et al. The BTK inhibitor ARQ 531 targets ibrutinib-resistant CLL and Richter transformation. Cancer Discov. 2018, 8, 1300–1315.
- Quinquenel, A.; Fornecker, L.M.; Letestu, R.; Ysebaert, L.; Fleury, C.; Lazarian, G.; Dilhuydy, M.S.; Nollet, D.; Guieze, R.; Feugier, P.; et al. Prevalence of BTK and PLCG2 mutations in a real-life CLL cohort still on ibrutinib after 3 years: A FILO group study. Blood 2019, 134, 641–644.
- Ran, F.; Liu, Y.; Wang, C.; Xu, Z.; Zhang, Y.; Liu, Y.; Zhao, G.; Ling, Y. Review of the development of BTK inhibitors in overcoming the clinical limitations of ibrutinib. Eur. J Med. Chem. 2022, 229, 114009.
- Woyach, J.A.; Furman, R.R.; Liu, T.M.; Ozer, H.G.; Zapatka, M.; Ruppert, A.S.; Xue, L.; Li, D.H.; Steggerda, S.M.; Versele, M.; et al. Re-sistance mechanisms for the Bruton’s tyrosine kinase inhibitor ibrutinib. N. Engl. J. Med. 2014, 370, 2286–2294.
- Woyach, J.A.; Ruppert, A.S.; Guinn, D.; Lehman, A.; Blachly, J.S.; Lozanski, A.; Heerema, N.A.; Zhao, W.; Coleman, J.; Jones, D.; et al. BTK C481S-mediated resistance to ibrutinib in chronic lymphocytic leukemia. J. Clin. Oncol. 2017, 35, 1437–1443.
- Woyach, J.; Huang, Y.; Rogers, K.; Bhat, S.A.; Grever, M.R.; Lozanski, A.; Doong, T.-J.; Blachly, J.S.; Lozanski, G.; Jones, D.; et al. Resistance to acalabrutinib in CLL is mediated primarily by BTK mutations. Blood 2019, 134, 504.
- George, B.; Chowdhury, S.M.; Hart, A.; Sircar, A.; Singh, S.K.; Nath, U.K.; Mamgain, M.; Singhal, N.K.; Sehgal, L.; Jain, N. Ibrutinib resistance mechanisms and treatment strategies for B-cell lymphomas. Cancers 2020, 12, 1328.
- Wang, H.; Zhang, W.; Yang, J.; Zhou, K. The resistance mechanisms and treatment strategies of BTK inhibitors in B-cell lymphoma. Hematol. Oncol. 2021, 39, 605–615.
- Bond, D.A.; Woyach, J.A. Targeting BTK in CLL: Beyond ibrutinib. Curr. Hematol. Malign. Rep. 2019, 14, 197–205.
- Jones, J.A.; Mato, A.R.; Wierda, W.G.; Davids, M.S.; Choi, M.; Cheson, B.D.; Furman, R.R.; Lamanna, N.; Barr, P.M.; Zhou, L.; et al. Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: An interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol. 2018, 19, 65–75.
- Skånland, S.S.; Mato, A.R. Overcoming resistance to targeted therapies in chronic lymphocytic leukemia. Blood Adv. 2021, 5, 334–343.
- Robinson, H.R.; Qi, J.; Cook, E.M.; Nichols, C.; Dadashian, E.L.; Underbayev, C.; Herman, S.E.M.; Saba, N.S.; Keyvanfar, K.; Sun, C.; et al. A CD19/CD3 bispecific antibody for effective immunotherapy of chronic lymphocytic leukemia in the ibrutinib era. Blood 2018, 132, 521–532.
- Turtle, C.J.; Hay, K.; Hanafi, L.-A.; Li, D.; Cherian, S.; Chen, X.; Wood, B.; Lozanski, A.; Byrd, J.C.; Heimfeld, S.; et al. Durable molecular remissions in chronic lymphocytic leukemia treated with CD19-specific chimeric antigen receptor–modified T cells after failure of ibrutinib. J. Clin. Oncol. 2017, 35, 3010–3020.
- Siddiqi, T.; Soumerai, J.D.; Dorritie, K.A.; Kathleen, A.; Dorritie, M.D.; Deborah, M.; Stephens, P.A.; Riedell, J.E.; Arnason, J.E.; Kipps, T.J.; et al. Rapid undetectable MRD (uMRD) responses in patients with relapsed/refractory (R/R) chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL/SLL) treated with lisocabtagene maraleucel (liso-cel), a CD19-directed CAR T cell product: Updated results from Transcend CLL 004, a phase 1/2 study including patients with high-risk disease previously treated with ibrutinib. Blood 2019, 134, 503.
- Sun, Y.; Zhao, X.; Ding, N.; Gao, H.; Wu, Y.; Yang, Y.; Zhao, M.; Hwang, J.; Song, Y.; Liu, W.; et al. PROTAC-induced BTK degradation as a novel therapy for mutated BTK C481S induced ibrutinib-resistant B-cell malignancies. Cell Res. 2018, 28, 779–781.
- Hallek, M.; Al-Sawaf, O. Chronic lymphocytic leukemia: 2022 update on diagnostic and therapeutic procedures. Am. J. Hematol. 2021, 96, 1679–1705.
- Smolewski, P.; Robak, T. Current treatment of refractory/relapsed chronic lymphocytic leukemia: A focus on novel drugs. Acta Haematol. 2020, 144, 365–379.
- O’Brien, S.M.; Brown, J.R.; Byrd, J.C.; Furman, R.R.; Ghia, P.; Sharman, J.P.; Wierda, W.G. Monitoring and managing BTK inhibitor treatment-related adverse events in clinical practice. Front. Oncol. 2021, 11, 720704.
- Von Hundelshausen, P.; Siess, W. Bleeding by Bruton tyrosine kinase-inhibitors: Dependency on drug type and disease. Cancers 2021, 13, 1103.
- Shatzel, J.J.; Olson, S.R.; Tao, D.L.; McCarty, O.J.T.; Danilov, A.V.; DeLoughery, T.G. Ibrutinib-associated bleeding: Patho-genesis, management and risk reduction strategies. J. Thromb. Haemost. 2017, 15, 835–847.
- Caron, F.; Leong, D.P.; Hillis, C.; Fraser, G.; Siegal, D. Current understanding of bleeding with ibrutinib use: A systematic review and meta-analysis. Blood Adv. 2017, 1, 772–778.
- Bye, A.P.; Unsworth, A.; Desborough, M.; Hildyard, C.; Appleby, N.; Bruce, D.; Kriek, N.; Nock, S.H.; Sage, T.; Hughes, C.; et al. Severe platelet dysfunction in NHL patients receiving ibrutinib is absent in patients receiving acalabrutinib. Blood Adv. 2017, 1, 2610–2623.
- Tam, C.S.; Dimopoulos, M.A.; Garcia-Sanz, R.; Trotman, J.; Opat, S.; Roberts, A.W.; Owen, R.G.; Song, Y.; Xu, W.; Zhu, J.; et al. Pooled safety analysis of zanubrutinib monotherapy in patients with B-cell malignancies. Blood Adv. 2021, 5, 2473–9529.
- Langerbeins, P.; Zhang, C.; Robrecht, S.; Cramer, P.; Fürstenau, M.; Al-Sawaf, O.; von Tresckow, J.; Fink, A.M.; Kreuzer, K.A.; Vehling-Kaiser, U.; et al. The CLL12 trial: Ibrutinib versus placebo in treatment-naïve, early stage chronic lymphocytic leu-kemia. Blood 2021, 10, 2021010845.
- Yun, S.; Vincelette, N.D.; Acharya, U.; Abraham, I. Risk of Atrial Fibrillation and Bleeding Diathesis Associated with Ibrutinib Treatment: A Systematic Review and Pooled Analysis of Four Randomized Controlled Trials. Clin. Lymphoma Myeloma Leuk. 2017, 17, 31–37.
- Mato, A.R.; Nabhan, C.; Barr, P.M.; Ujjani, C.S.; Hill, B.T.; Lamanna, N.; Skarbnik, A.P.; Howlett, C.; Pu, J.J.; Sehgal, A.R.; et al. Outcomes of CLL patients treated with sequential kinase inhibitor therapy: A real world experience. Blood 2016, 128, 199–2205.
- Leong, D.P.; Caron, F.; Hillis, C.; Duan, A.; Healey, J.S.; Fraser, G.; Siegal, D. The risk of atrial fibrillation with ibrutinib use: A systematic review and meta-analysis. Blood 2016, 128, 138–140.
- Guha, A.; Derbala, M.H.; Zhao, Q.; Wiczer, T.E.; Woyach, J.A.; Byrd, J.C.; Awan, F.T.; Addison, D. Ventricular arrhythmias following ibrutinib initiation for lymphoid malignancies. J. Am. Coll. Cardiol. 2018, 72, 697–698.
- Grewal, U.S.; Thotamgari, S.R.; Sheth, A.R.; Gaddam, S.J.; Ahmad, J.; Beedupalli, K.; Dominic, P. Cardiovascular complications associated with novel agents in the chronic lymphocytic leukemia armamentarium: A pharmacovigilance analysis. Int. J. Cardiol. 2021, 344, 186–189.
- Caldeira, D.; Alves, D.; Costa, J.; Ferreira, J.J.; Pinto, F.J. Ibrutinib increases the risk of hypertension and atrial fibrillation: Systematic review and meta-analysis. PLoS ONE 2019, 14, e0211228.
- Binsah, G.; Philip, T.A.; Ferrajoli, A.; Jan, B.; Jain, N.; Wierda, W.G.; O’Brien, S.; Durand, J.B.; Keating, M.J. An observational study of the occurrence of atrial fibrillation and hypertension in patientstreated with ibrutinib. Blood 2014, 124, 5657.
- O’Brien, S.; Hillmen, P.; Coutre, S.; Barr, P.M.; Fraser, G.; Tedeschi, A.; Burger, J.A.; Dilhuydy, M.S.; Hess, G.; Moreno, C.; et al. Safety analysis of four randomized controlled studies of ibrutinib in patients with chronic lymphocytic leukemia/small lymphocytic lymphoma or mantle cell lymphoma. Clin. Lymphoma Myeloma Leuk. 2018, 18, 648–657.
- Dickerson, T.; Wiczer, T.; Waller, A.; Philippon, J.; Porter, K.; Haddad, D.; Guha, A.; Rogers, K.A.; Bhat, S.; Byrd, J.C.; et al. Hypertension and incident cardiovascular events following ibrutinib initiation. Blood 2019, 134, 1919–1928.
- Vitale, C.; Salvetti, C.; Griggio, V.; Porrazzo, M.; Schiattone, L.; Zamprogna, G.; Visentin, A.; Vassallo, F.; Cassin, R.; Rigolin, G.M.; et al. Preexisting and treatment-emergent autoimmune cytopenias in patients with CLL treated with targeted drugs. Blood 2021, 137, 3507–3517.
- Noto, A.; Cassin, R.; Mattiello, V.; Reda, G. The role of novel agents in treating CLL-associated autoimmune hemolytic anemia. J. Clin. Med. 2021, 10, 2064.
- Ye, B.; Zhou, C.; Guo, H.; Zheng, M. Effects of BTK signalling in pathogenic microorganism infections. J. Cell. Mol. Med. 2019, 23, 6522.
- McDonald, C.; Xanthopoulos, C.; Kostareli, E. The role of Bruton’s tyrosine kinase in the immune system and disease. Immunology 2021, 164, 722–736.
- Pleyer, C.; Sun, C.; Desai, S.; Ahn, I.E.; Tian, X.; Nierman, P.; Soto, S.; Superata, J.; Valdez, J.; Lotter, J.; et al. Reconsti-tution of humoral immunity and decreased risk of infections in patients with chronic lymphocytic leukemia treated with Bruton tyrosine kinase inhibitors. Leuk. Lymphoma 2020, 61, 2375–2382.
- Varughese, T.; Taur, Y.; Cohen, N.; Palomba, M.L.; Seo, S.K.; Hohl, T.M.; Redelman-Sidi, G. Serious Infections in Patients Receiving Ibrutinib for Treatment of Lymphoid Cancer. Clin. Infect. Dis. 2018, 67, 687–692.
- Holowka, T.; Cheung, H.; Malinis, M.; Gan, G.; Deng, Y.; Perreault, S.; Isufi, I.; Azar, M.M. Incidence and associated risk factors for invasive fungal infections and other serious infections in patients on ibrutinib. J. Infect. Chemother. 2021, 27, 1700–1705.
- Frei, M.; Aitken, S.L.; Jain, N.; Thompson, P.; Wierda, W.; Kontoyiannis, D.P.; DiPippo, A.J. Incidence and characterization of fungal infections in chronic lymphocytic leukemia patients receiving ibrutinib. Leuk. Lymphoma 2020, 61, 2488–2491.
- Mauro, F.; Giannarelli, D.; Visentin, A.; Reda, G.; Sportoletti, P.; Frustaci, A.; Chiarenza, A.; Ciolli, S.; Vitale, C.; Laurenti, L.; et al. Prognostic impact and risk factors of infections in patients with chronic lymphocytic leukemia treated with ibrutinib. Cancers 2021, 13, 3240.
- Lipsky, A.; Lamanna, N. Managing toxicities of Bruton tyrosine kinase inhibitors. Hematology 2020, 2020, 336–345.
- Pleyer, C.; Ali, M.A.; Cohen, J.I.; Tian, X.; Soto, S.; Ahn, I.E.; Gaglione, E.M.; Nierman, P.; Marti, G.E.; Hesdorffer, C.; et al. Effect of Bruton tyrosine kinase inhibitor on efficacy of adjuvanted recombinant hepatitis B and zoster vaccines. Blood 2021, 137, 185–189.
- Rhodes, J.M.; LoRe, V.A., 3rd; Mato, A.R.; Chong, E.A.; Barrientos, J.C.; Gerson, J.N.; Barta, S.K.; Landsburg, D.J.; Nasta, S.D.; Svoboda, J.; et al. Ibrutinib-associated arthralgias/myalgias in patients with chronic lymphocytic leu-kemia: Incidence and impact on clinical outcomes. Clin. Lymphoma Myeloma Leukemia 2020, 20, 438–444.
- Roeker, L.E.; Eyre, T.A.; Thompson, M.C.; Lamanna, N.; Coltoff, A.R.; Davids, M.S.; Baker, P.O.; Leslie, L.A.; Rogers, K.A.; Allan, J.N.; et al. Outcomes of front-line ibrutinib treated CLL patients excluded from landmark clinical trial. Am. J. Hematol. 2021, 138, 1768–1773.
- Mato, A.; Jahnke, J.; Li, P.; Mehra, M.; Ladage, V.P.; Mahler, M.; Huntington, S.; Doshi, J.A. Real-world treatment and outcomes among older adults with chronic lymphocytic leukemia before the novel agents era. Haematologica 2018, 103, e462–e465.
- Forum, U.C. Ibrutinib for relapsed/refractory chronic lymphocytic leukemia: A UK and Ireland analysis of outcomes in 315 patients. Haematologica 2016, 101, 1563–1572.
- Stephens, D.M.; Byrd, J.C. How I manage ibrutinib intolerance and complications in patients with chronic lymphocytic leu-kemia. Blood 2019, 133, 1298–1307.
- Awan, F.T.; Schuh, A.; Brown, J.R.; Furman, R.R.; Pagel, J.M.; Hillmen, P.; Stephens, D.M.; Woyach, J.; Bibikova, E.; Charuworn, P.; et al. Acalabrutinib monotherapy in patients with chronic lymphocytic leukemia who are intolerant to ibrutinib. Blood Adv. 2019, 3, 1553–1562.
- Iberri, D.J.; Kwong, B.Y.; Stevens, L.A.; Coutre, S.E.; Kim, J.; Sabile, J.M.; Advani, R.H. Ibrutinib-associated rash: A single-centre experience of clinicopathological features and management. Br. J. Haematol. 2016, 180, 164–166.
- Sibaud, V.; Beylot-Barry, M.; Protin, C.; Vigarios, E.; Recher, C.; Ysebaert, L. Dermatological Toxicities of Bruton’s Tyrosine Kinase Inhibitors. Am. J. Clin. Dermatol. 2020, 21, 799–812.
- Bitar, C.; Farooqui, M.Z.H.; Valdez, J.; Saba, N.S.; Soto, S.; Bray, A.; Marti, G.; Wiestner, A.; Cowen, E.W. Hair and nail changes during long-term therapy with ibrutinib for chronic lymphocytic leukemia. JAMA Dermatol. 2016, 152, 698–701.
- Abbas, H.A.; Wierda, W.G. Acalabrutinib: A Selective Bruton Tyrosine Kinase Inhibitor for the Treatment of B-Cell Malignancies. Front. Oncol. 2021, 11, 668162.
- Sun, C.C.L.; Nierman, P.K.; Kendall, E.K.; Cheung, J.; Gulrajani, M.; Herman, S.E.M.; Pleyer, C.; Ahn, I.E.; Stetler-Stevenson, M.; Yuan, C.M.; et al. Clinical and biological implications of target occupancy in CLL treated with the BTK inhibitor acalabrutinib. Blood 2020, 136, 93–105.