Acute myeloid leukemia (AML) is a clonal disorder that affects myeloid progenitor cells residing in the bone marrow (BM). This implies altered differentiation with subsequent abnormal proliferation and accumulation of inadequately matured myeloid cells. The detection of leukemic cells moved in the last two decades from immune-phenotyping to polymerase chain reaction (PCR) and real-time quantitative PCR (RT-qPCR). This technique was shown to be reproducible, accurate and highly sensitive for MRD monitoring, with a significant capacity in predicting prognosis, treatment effectiveness and relapse risk. NGS or massively parallel sequencing is a revolutionary method of DNA and RNA sequencing. It is called parallel because it sequences millions of DNA fragments simultaneously. In the early years of its appearance, Next Generation Sequencing (NGS) platforms were used primarily for cancer research purposes. Recently, they are increasingly emerging as irreplaceable diagnostic tools in clinical settings.
- acute myeloid leukemia (AML)
- RT-qPCR
- MDR
- digital droplet PCR (ddPCR)
- next-generation sequencing (NGS)
- metabolomics analysis
- metabolomic profiling
1. Introduction
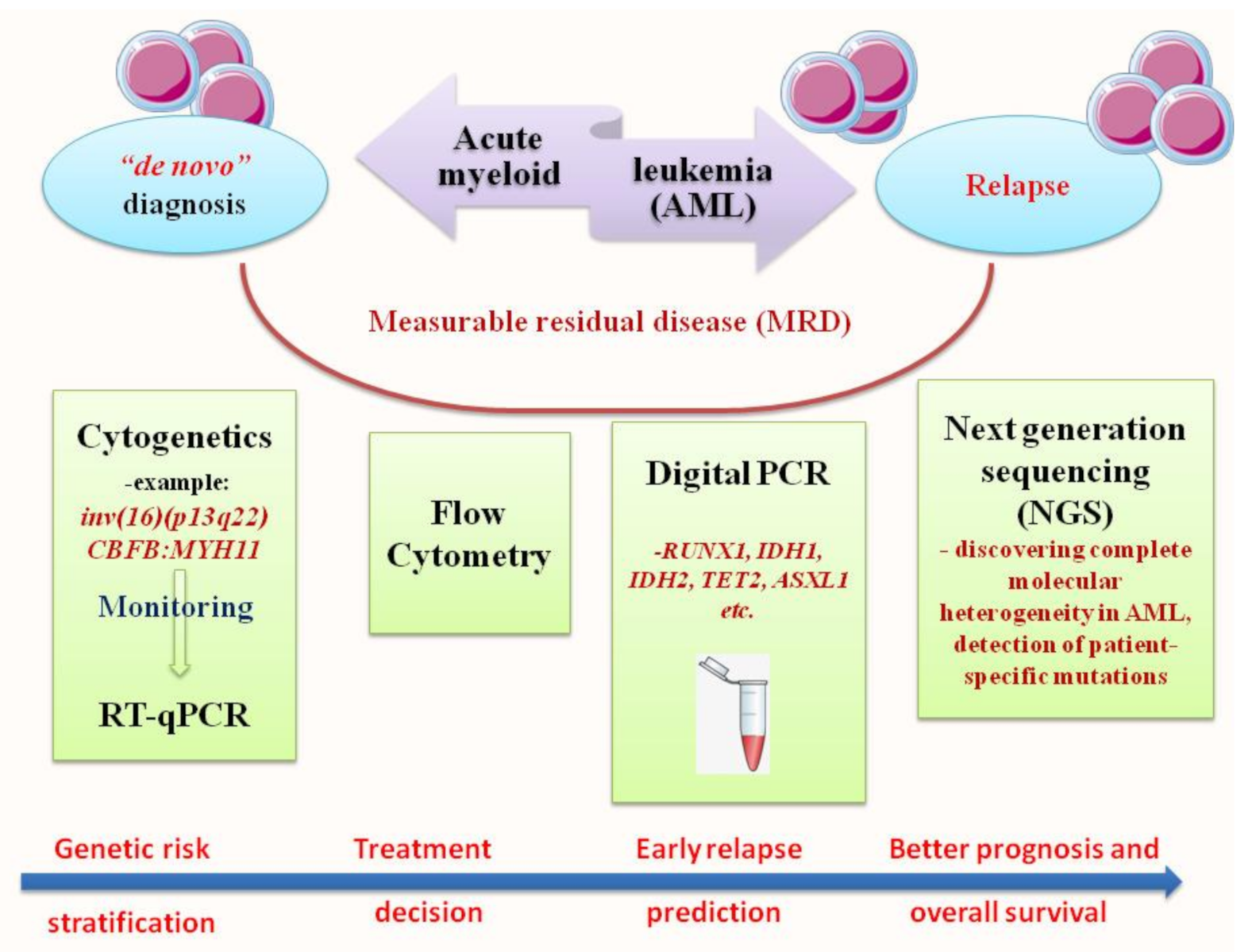
2. RT-qPCR: The Gold Standard for Diagnosis and Prognosis Stratification in AML
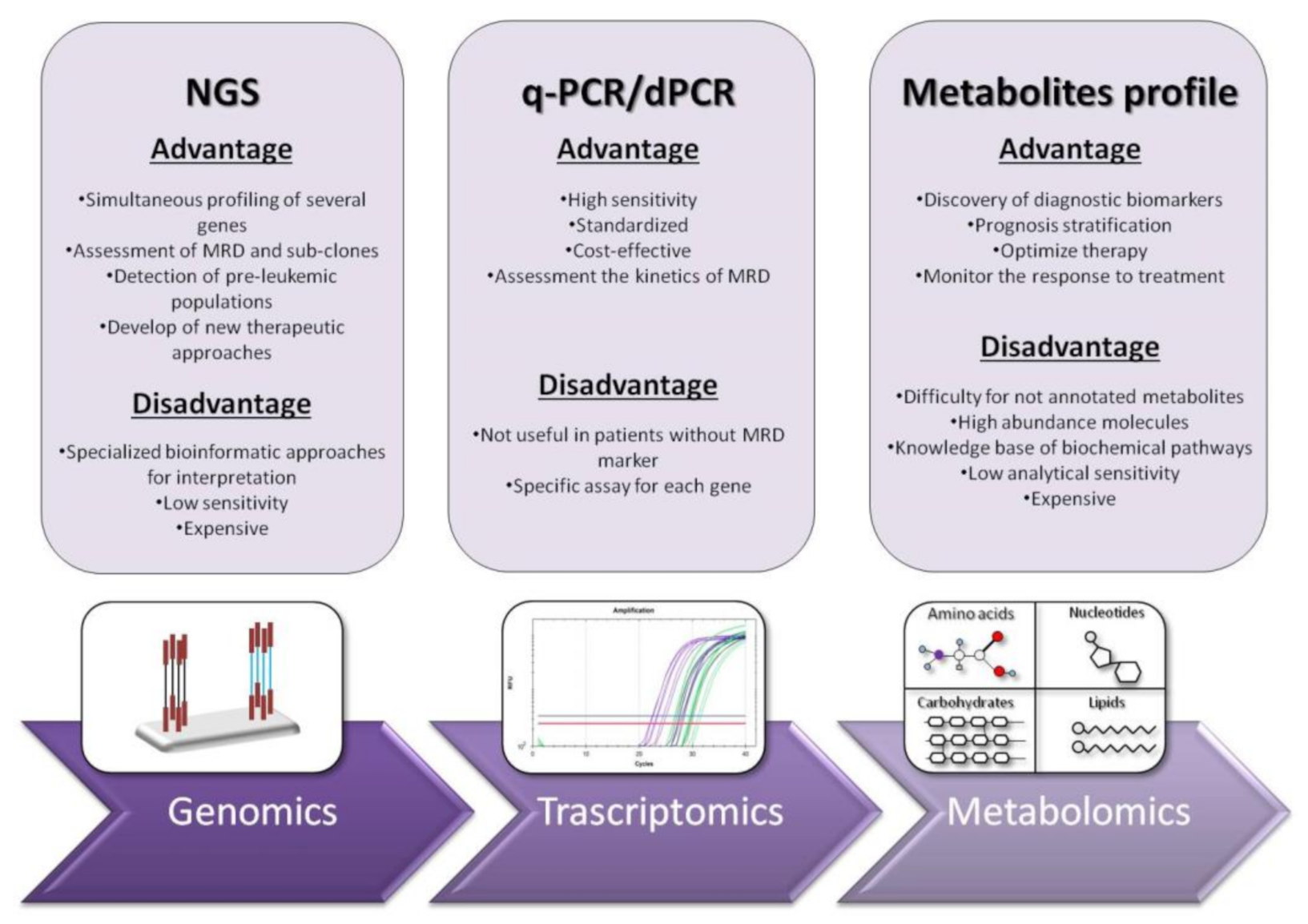
3. Next Generation Sequencing (NGS): Unveiling of the Molecular Landscape in Myeloid Neoplasms
This entry is adapted from the peer-reviewed paper 10.3390/jcm11030483
References
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441.
- Grove, C.S.; Vassiliou, G.S. Acute myeloid leukaemia: A paradigm for the clonal evolution of cancer? Dis. Models Mech. 2014, 7, 941–951.
- Goel, H.; Rahul, E.; Gupta, I.; Chopra, A.; Ranjan, A.; Gupta, A.K.; Meena, J.P.; Viswanathan, G.K.; Bakhshi, S.; Misra, A. Molecular and genomic landscapes in secondary & therapy related acute myeloid leukemia. Am. J. Blood Res. 2021, 11, 472.
- Höllein, A.; Nadarajah, N.; Meggendorfer, M.; Jeromin, S.; Kern, W.; Haferlach, C.; Haferlach, T. Molecular characterization of aml with runx1-runx1t1 at diagnosis and relapse reveals net loss of co-mutations. HemaSphere 2019, 3, e178.
- Liquori, A.; Ibañez, M.; Sargas, C.; Sanz, M.Á.; Barragán, E.; Cervera, J. Acute promyelocytic leukemia: A constellation of molecular events around a single pml-rara fusion gene. Cancers 2020, 12, 624.
- Yang, J.J.; Park, T.S.; Wan, T.S. Recurrent cytogenetic abnormalities in acute myeloid leukemia. Cancer Cytogenet. 2017, 1541, 223–245.
- Cancer Genome Atlas Research Network. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074.
- Kansal, R. Toward integrated genomic diagnosis in routine diagnostic pathology by the world health organization classification of acute myeloid leukemia. J. Clin. Haematol. 2020, 1, 2.
- Carter, J.L.; Hege, K.; Yang, J.; Kalpage, H.A.; Su, Y.; Edwards, H.; Hüttemann, M.; Taub, J.W.; Ge, Y. Targeting multiple signaling pathways: The new approach to acute myeloid leukemia therapy. Signal Transduct. Target. Ther. 2020, 5, 288.
- Calabrese, C.; Panuzzo, C.; Stanga, S.; Andreani, G.; Ravera, S.; Maglione, A.; Pironi, L.; Petiti, J.; Shahzad Ali, M.; Scaravaglio, P. Deferasirox-dependent iron chelation enhances mitochondrial dysfunction and restores p53 signaling by stabilization of p53 family members in leukemic cells. Int. J. Mol. Sci. 2020, 21, 7674.
- Panuzzo, C.; Signorino, E.; Calabrese, C.; Ali, M.S.; Petiti, J.; Bracco, E.; Cilloni, D. Landscape of tumor suppressor mutations in acute myeloid leukemia. J. Clin. Med. 2020, 9, 802.
- Saliba, A.N.; John, A.J.; Kaufmann, S.H. Resistance to venetoclax and hypomethylating agents in acute myeloid leukemia. Cancer Drug Resist. 2021, 4, 125.
- Liu, X.; Gong, Y. Isocitrate dehydrogenase inhibitors in acute myeloid leukemia. Biomark. Res. 2019, 7, 22.
- Sekeres, M.A.; Guyatt, G.; Abel, G.; Alibhai, S.; Altman, J.K.; Buckstein, R.; Choe, H.; Desai, P.; Erba, H.; Hourigan, C.S. American society of hematology 2020 guidelines for treating newly diagnosed acute myeloid leukemia in older adults. Blood Adv. 2020, 4, 3528–3549.
- Voso, M.T.; Ottone, T.; Lavorgna, S.; Venditti, A.; Maurillo, L.; Lo-Coco, F.; Buccisano, F. Mrd in aml: The role of new techniques. Front. Oncol. 2019, 9, 655.
- Schuurhuis, G.J.; Heuser, M.; Freeman, S.; Béné, M.-C.; Buccisano, F.; Cloos, J.; Grimwade, D.; Haferlach, T.; Hills, R.K.; Hourigan, C.S. Minimal/measurable residual disease in aml: A consensus document from the european leukemianet mrd working party. Blood J. Am. Soc. Hematol. 2018, 131, 1275–1291.
- Hauwel, M.; Matthes, T. Minimal residual disease monitoring: The new standard for treatment evaluation of haematological malignancies? Swiss Med. Wkly. 2014, 144, w13907.
- Gabert, J.; Beillard, E.; van der Velden, V.H.J.; Bi, W.; Grimwade, D.; Pallisgaard, N.; Barbany, G.; Cazzaniga, G.; Cayuela, J.M.; Cavé, H.; et al. Standardization and quality control studies of ‘real-time’ quantitative reverse transcriptase polymerase chain reaction of fusion gene transcripts for residual disease detection in leukemia—A Europe against cancer program. Leukemia 2003, 17, 2318–2357.
- Aitken, M.J.; Ravandi, F.; Patel, K.P.; Short, N.J. Prognostic and therapeutic implications of measurable residual disease in acute myeloid leukemia. J. Hematol. Oncol. 2021, 14, 137.
- Ossenkoppele, G.; Schuurhuis, G.J. MRD in AML: Does it already guide therapy decision-making? Hematol. 2014 Am. Soc. Hematol. Educ. Program Book 2016, 2016, 356–365.
- Döhner, H.; Estey, E.; Grimwade, D.; Amadori, S.; Appelbaum, F.R.; Büchner, T.; Dombret, H.; Ebert, B.L.; Fenaux, P.; Larson, R.A. Diagnosis and management of aml in adults: 2017 eln recommendations from an international expert panel. Blood J. Am. Soc. Hematol. 2017, 129, 424–447.
- Chendamarai, E.; Balasubramanian, P.; George, B.; Viswabandya, A.; Abraham, A.; Ahmed, R.; Alex, A.A.; Ganesan, S.; Lakshmi, K.M.; Sitaram, U. Role of minimal residual disease monitoring in acute promyelocytic leukemia treated with arsenic trioxide in frontline therapy. Blood J. Am. Soc. Hematol. 2012, 119, 3413–3419.
- Chen, Z.; Tong, Y.; Li, Y.; Gao, Q.; Wang, Q.; Fu, C.; Xia, Z. Development and validation of a 3-plex rt-qpcr assay for the simultaneous detection and quantitation of the three pml-rara fusion transcripts in acute promyelocytic leukemia. PLoS ONE 2015, 10, e0122530.
- Willekens, C.; Blanchet, O.; Renneville, A.; Cornillet-Lefebvre, P.; Pautas, C.; Guieze, R.; Ifrah, N.; Dombret, H.; Jourdan, E.; Preudhomme, C. Prospective long-term minimal residual disease monitoring using rq-pcr in runx1-runx1t1-positive acute myeloid leukemia: Results of the french cbf-2006 trial. Haematologica 2016, 101, 328.
- Jourdan, E.; Boissel, N.; Chevret, S.; Delabesse, E.; Renneville, A.; Cornillet, P.; Blanchet, O.; Cayuela, J.-M.; Recher, C.; Raffoux, E. Prospective evaluation of gene mutations and minimal residual disease in patients with core binding factor acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2013, 121, 2213–2223.
- Puckrin, R.; Atenafu, E.G.; Claudio, J.O.; Chan, S.; Gupta, V.; Maze, D.; McNamara, C.; Murphy, T.; Schuh, A.C.; Yee, K. Measurable residual disease monitoring provides insufficient lead-time to prevent morphological relapse in the majority of patients with core-binding factor acute myeloid leukemia. Haematologica 2021, 106, 56–63.
- Falini, B.; Brunetti, L.; Sportoletti, P.; Martelli, M.P. Npm1-mutated acute myeloid leukemia: From bench to bedside. Blood 2020, 136, 1707–1721.
- Gorello, P.; Cazzaniga, G.; Alberti, F.; Dell’Oro, M.; Gottardi, E.; Specchia, G.; Roti, G.; Rosati, R.; Martelli, M.; Diverio, D. Quantitative assessment of minimal residual disease in acute myeloid leukemia carrying nucleophosmin (npm1) gene mutations. Leukemia 2006, 20, 1103–1108.
- Forghieri, F.; Comoli, P.; Marasca, R.; Potenza, L.; Luppi, M. Minimal/measurable residual disease monitoring in npm1-mutated acute myeloid leukemia: A clinical viewpoint and perspectives. Int. J. Mol. Sci. 2018, 19, 3492.
- Tiong, S.; Dillon, R.; Ivey, A.; Kok, C.H.; Kuzich, J.A.; Thiagarajah, N.; Bajel, A.; Potter, N.; Smith, M.; Hemmaway, C. The natural history of npm1mut measurable residual disease (MRD) positivity after completion of chemotherapy in acute myeloid leukemia (AML). Blood 2020, 136, 25–27.
- Lussana, F.; Caprioli, C.; Stefanoni, P.; Pavoni, C.; Spinelli, O.; Buklijas, K.; Michelato, A.; Borleri, G.; Algarotti, A.; Micò, C. Molecular detection of minimal residual disease before allogeneic stem cell transplantation predicts a high incidence of early relapse in adult patients with npm1 positive acute myeloid leukemia. Cancers 2019, 11, 1455.
- Balsat, M.; Renneville, A.; Thomas, X.; de Botton, S.; Caillot, D.; Marceau, A.; Lemasle, E.; Marolleau, J.-P.; Nibourel, O.; Berthon, C. Postinduction minimal residual disease predicts outcome and benefit from allogeneic stem cell transplantation in acute myeloid leukemia with npm1 mutation: A study by the acute leukemia french association group. J. Clin. Oncol. 2017, 35, 185–193.
- Behjati, S.; Tarpey, P.S. What is next generation sequencing? Arch. Dis. Child. Educ. Pract. 2013, 98, 236–238.
- Anderson, M.W.; Schrijver, I. Next generation DNA sequencing and the future of genomic medicine. Genes 2010, 1, 38–69.
- Bacher, U.; Shumilov, E.; Flach, J.; Porret, N.; Joncourt, R.; Wiedemann, G.; Fiedler, M.; Novak, U.; Amstutz, U.; Pabst, T. Challenges in the introduction of next-generation sequencing (ngs) for diagnostics of myeloid malignancies into clinical routine use. Blood Cancer J. 2018, 8, 113.
- Vainchenker, W.; Kralovics, R. Genetic basis and molecular pathophysiology of classical myeloproliferative neoplasms. Blood J. Am. Soc. Hematol. 2017, 129, 667–679.
- Vannucchi, A.M.; Lasho, T.; Guglielmelli, P.; Biamonte, F.; Pardanani, A.; Pereira, A.; Finke, C.; Score, J.; Gangat, N.; Mannarelli, C. Mutations and prognosis in primary myelofibrosis. Leukemia 2013, 27, 1861–1869.
- Tefferi, A.; Lasho, T.L.; Guglielmelli, P.; Finke, C.M.; Rotunno, G.; Elala, Y.; Pacilli, A.; Hanson, C.A.; Pancrazzi, A.; Ketterling, R.P. Targeted deep sequencing in polycythemia vera and essential thrombocythemia. Blood Adv. 2016, 1, 21–30.
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N. Genomic classification and prognosis in acute myeloid leukemia. N. Engl. J. Med. 2016, 374, 2209–2221.
- Thol, F.; Kölking, B.; Damm, F.; Reinhardt, K.; Klusmann, J.H.; Reinhardt, D.; von Neuhoff, N.; Brugman, M.H.; Schlegelberger, B.; Suerbaum, S. Next-generation sequencing for minimal residual disease monitoring in acute myeloid leukemia patients with flt3-itd or npm1 mutations. Genes Chromosomes Cancer 2012, 51, 689–695.
- Morita, K.; Kantarjian, H.M.; Wang, F.; Yan, Y.; Bueso-Ramos, C.; Sasaki, K.; Issa, G.C.; Wang, S.; Jorgensen, J.; Song, X. Clearance of somatic mutations at remission and the risk of relapse in acute myeloid leukemia. J. Clin. Oncol. 2018, 36, 1788.
- Kohlmann, A.; Nadarajah, N.; Alpermann, T.; Grossmann, V.; Schindela, S.; Dicker, F.; Roller, A.; Kern, W.; Haferlach, C.; Schnittger, S. Monitoring of residual disease by next-generation deep-sequencing of runx1 mutations can identify acute myeloid leukemia patients with resistant disease. Leukemia 2014, 28, 129–137.
- Jongen-Lavrencic, M.; Grob, T.; Hanekamp, D.; Kavelaars, F.G.; Al Hinai, A.; Zeilemaker, A.; Erpelinck-Verschueren, C.A.; Gradowska, P.L.; Meijer, R.; Cloos, J. Molecular minimal residual disease in acute myeloid leukemia. N. Engl. J. Med. 2018, 378, 1189–1199.
- Mertens, F.; Johansson, B.; Fioretos, T.; Mitelman, F. The emerging complexity of gene fusions in cancer. Nat. Rev. Cancer 2015, 15, 371–381.
- Leisch, M.; Jansko, B.; Zaborsky, N.; Greil, R.; Pleyer, L. Next generation sequencing in aml—On the way to becoming a new standard for treatment initiation and/or modulation? Cancers 2019, 11, 252.
- Metzeler, K.H.; Herold, T.; Rothenberg-Thurley, M.; Amler, S.; Sauerland, M.C.; Görlich, D.; Schneider, S.; Konstandin, N.P.; Dufour, A.; Bräundl, K. Spectrum and prognostic relevance of driver gene mutations in acute myeloid leukemia. Blood J. Am. Soc. Hematol. 2016, 128, 686–698.
- Bhatnagar, B.; Eisfeld, A.K.; Nicolet, D.; Mrózek, K.; Blachly, J.S.; Orwick, S.; Lucas, D.M.; Kohlschmidt, J.; Blum, W.; Kolitz, J.E. Persistence of dnmt 3a r882 mutations during remission does not adversely affect outcomes of patients with acute myeloid leukaemia. Br. J. Haematol. 2016, 175, 226–236.
- Zebisch, A.; Lal, R.; Müller, M.; Lind, K.; Kashofer, K.; Girschikofsky, M.; Fuchs, D.; Wölfler, A.; Geigl, J.B.; Sill, H. Acute myeloid leukemia with tp53 germ line mutations. Blood J. Am. Soc. Hematol. 2016, 128, 2270–2272.