Epinephrine (E) and norepinephrine (NE) play diverse roles in our body’s physiology. In addition to their role in the peripheral nervous system (PNS), E/NE systems including their receptors are critical to the central nervous system (CNS) and to mental health.
- Radiotracer
- adrenergic receptor
- positron emission tomography
- fluorine-18
- radiolabelling
- organic synthesis
Note:All the information in this draft can be edited by authors. And the entry will be online only after authors edit and submit it.
1. Introduction
Positron emission tomography (PET) is a noninvasive and highly sensitive in vivo imaging technique that uses small amounts of radiotracers to detect the concentration of relevant biomarkers in tissues such as receptors, enzymes, and transporters. These radiotracers can be used to characterize neurochemical changes in neuropsychiatric diseases and also to measure drug pharmacokinetics and pharmacodynamics directly in the human body including the brain [1,2]. PET technology requires positron-emitting radioisotopes, a radiotracer synthesis unit, a PET scanner, and data acquisition components. Among the PET isotopes, cyclotron produced carbon-11 (C-11, t1/2 = 20.34 min), nitrogen-13 (N-13, t1/2 = 9.96 min), and fluorine-18 (F-18, t1/2 = 109.77 min) are frequently used for PET-neuroimaging. PET imaging is particularly valuable to characterize investigational drugs and their target proteins, providing a valuable tool for clinical neuroscience [3].
PET radiotracers for the central nervous system (CNS) should have proper pharmacokinetic profile in the brain and to achieve this property, they were designed to meet five molecular properties: a) molecular weight <500 kDa, b) Log D7.4 between ~1 to 3 (lipophilicity factor), c) number of hydrogen bond donors <5, d) number of hydrogen bond acceptors <10, and e) topological polar surface area <90 Å2.
2. α1-AR PET Radiotracers
Three highly homologous subunits of α-1 ARs, α1A, α1B, and α1D, have been shown to have different amino acid sequence, pharmacological properties, and tissue distributions [58–60]. A detailed review of α1-AR pharmacology is given by Michael and Perez [58]. Binding of NE/E to any of the α1-AR subtypes is stimulatory and activates Gq/11-signalling pathway, which involves phospholipase C activation, generation of secondary messengers, inositol triphosphate and diacyl glycerol and intracellular calcium mobilization. In PNS, as well as in the brain’s vascular system it results in smooth muscle contraction and vasoconstriction. While the signaling effects of α1-ARs in the cardiovascular system are well studied [61,62], the role of α1-ARs in CNS is complex and not completely understood. The Bmax values of α-1 ARs were measured from saturation assays using [3H]prazosin (a selective α-1 blocker) with tissue homogenates from rats and the observed binding capacities (fmol/mg tissue) of prazosin in cortex, hippocampus, and cerebellum were 14.49 ± 0.38, 11.03 ± 0.39, and 7.72 ± 0.11, respectively [23,63]. The α1-ARs are postsynaptic receptors and can also modulate release of NE. In the human brain, α1-AR subtypes are localized in amygdala, cerebellum, thalamus, hippocampus, and to some extent in striatum [20,22,64].
The anti-depressant effects of noradrenergic enhancing drugs as well as their effects on anxiety and stress reactivity points to their relevance of α1A, α1D to these behaviors [21,28,65]. Furthermore, α1A-ARs regulate GABAergic and NMDAergic neurons [20]. A decrease in α1A-AR mRNA expression was observed in prefrontal cortex of subjects with dementia [66,67]. The α1B-ARs has also been shown to be crucial to brain function and disease [18]. For example, α1B-knockout (KO) mice study revealed that α1B-AR modulates behavior, showing increased reaction to novel situations [24,27]. In addition, the locomotor and rewarding effects of psychostimulants and opiates were decreased in α1B-KO mice, highlighting their role in the pharmacological effects of these drugs [24]. On the other hand, studies using the α1B-overexpression model suggest their involvement in neurodegenerative diseases [25].
Several α1-AR agonists and antagonists (α1-blockers) are available in the market as drugs to treat various heart and brain disorders [68,69]. Pharmacology of most of these drugs is complicated by the fact that they have strong affinities for other receptor systems, such as serotonin and dopamine receptors. The need to develop α1-AR selective/α1-AR subtype specific drugs is demanding. Undoubtedly, PET radiotracers selective for α1-AR are valuable to assess α1-ARs contribution to brain function and disease. In the late 1980s, prazosin, used for the treatment of hypertension, was labelled with carbon-11 to image α1-ARs with PET [49]. Following this, [11C]prazosin analogous, [11C]bunazosin and [11C]GB67 (Figure 1A) were developed as PET radiotracers to image α1-ARs in the cardiovascular system [49,70]. These tracers were shown to have limited BBB permeability and were deemed to be not suitable for PET-neuroimaging. Efforts to develop PET radiotracers to image α1-ARs in the CNS was mainly based on antipsychotic drugs such as clozapine, sertindole, olanzapine, and risperidone that have mixed binding affinities for D2, 5HT2A receptors and α1-ARs in the nanomolar range. Their affinity for α1-ARs has been shown to contribute to antipsychotic efficacy uncovering their role in psychoses [71,72].
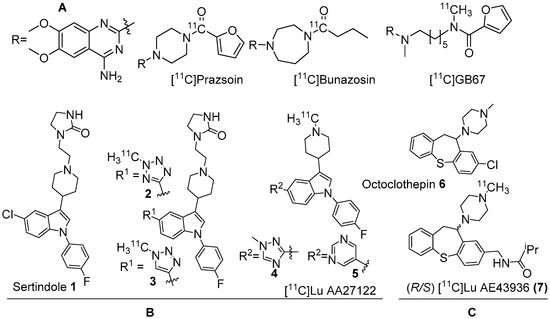
Figure 1. (A) Earlier PET radiotracers, [11C]Prazosin, [11C]Bunazosin, and [11C]GB67 for cardiac α1-AR imaging. (B) Antagonist PET radiotracers based on sertindole. (C) Antagonist PET radiotracers based on octoclothepin for brain α1-AR imaging.
Two C-11 labelled analogs of the atypical antipsychotic drug sertindole were first reported by Balle et al. in the early 2000s (Figure 1B) [73,74]. Several other analogs with good affinities (Ki < 10 nM) for α1-ARs were subsequently reported by the same group. Two analogs, in which chlorine in sertindole (1) is replaced by a 2-methyl-tetrazol-5-yl (2) and a 1-methyl-1,2,3-triazol-4-yl (3), were labelled with C-11 [73,74]. The in vitro affinities (Ki) of 1, 2 and 3 for α1-ARs are 1.4, 1.8 and 9.5 nM, respectively. Both [11C]2 and [11C]3 were prepared using 11C-methylation with [11C]methyl triflate from their corresponding N-desmethyl precursors. The molar activity of [11C]2 and [11C]3 was reported at 70 GBq/μmol and 15 GBq/μmol, respectively. Their brain distribution examined with PET in Cynomolgus monkeys showed that brain uptake of [11C]2 and [11C]3 was slow and low, with [11C]3 showing somewhat higher brain uptake than [11C]2. Their brain distribution was homogenous and specific binding to α1-ARs could not be demonstrated. It was also concluded that these two radiotracers were not suitable to image α1-ARs in brain owing to rapid metabolism, substantial distribution to other organs, and substrates for active efflux mechanism.
3. β-ARs and Nonselective PET Radiotracers
β-ARs are associated with Gs-heterotrimeric G-protein and mediate intracellular signaling through adenyl cyclase activation and cyclic adenosine monophosphate (cAMP) production. β-ARs
are classified into β1, β2, and β3 subtypes, in which, the former two have been much more explored [42,46]. Quite a lot of selective and nonselective β-AR agonists and antagonists (blockers) are available as drugs in the market to treat various cardiac and pulmonary disorders. In the brain, β-ARs are localized in the frontal cortex, striatum, thalamus, putamen, amygdala, cerebellum and hippocampus [48]. The density of β-ARs in brain is sensitive to brain pathophysiology. Notably, the density of β-ARs decrease with age [40]. Light microscopic autoradiography using [3H]dihydroaloprenolol (a selective β-blocker) with rat brain sections has shown a wide distribution of β-ARs in forebrain and cerebellum regions (Bmax = 23 fmol/mg tissue) [103]. Similarly, Bmax value of 18 fmol/mg protein was reported in pre-frontal cortex of subjects with Parkinson’s disease [48,104]. By altering the Ca2+ levels through N-methyl-D-aspartate (NMDA) receptors in hippocampus, β-ARs modulate synaptic plasticity, including that needed for memory [44,45]. The blockade of β-ARs is associated with a small increased risk for Alzheimer’s and Parkinson’s disease [43,105]. In addition, abnormal function and densities of β-ARs have been reported in mood disorders and schizophrenia [41,47,106].
Several radioligands, mostly based on β-blockers, were validated for imaging of β-ARs in the heart [50]. The majority of β-blockers possess a hydroxyl propylamine moiety in their structures that is vital for binding to β-ARs and this moiety was maintained in most of these radioligands. PET radiotracers have succeeded in imaging and quantifying myocardial and pulmonary β-ARs in human [107,108], whereas, PET radiotracers for cerebral β-ARs have been more challenging. The clinical PET radiotracers for cardiac β-ARs have negative Log P values (<−2), which is not suitable for imaging the brain. Several lipophilic and high to moderate affinity β-AR nonselective antagonists were explored as PET radiotracers to image β-ARs in the brain.
In 2001, Fazio’s group described two isomers (R/S) of C-11 labelled bisprolol (β1 Ki 1.6 nM, β2 Ki 100 nM and Log P7.4 = −0.2) (26, Figure 2) to image β1-ARs in the brain [117]. The radiosynthesis of 26 was accomplished via reductive amination using [11C]acetone and desisopropyl bisprolol precursor with 129.5 ± 37 GBq/μmol molar activity at the EOS. They observed little specific uptake of 26 in β1-AR rich regions in the rat’s brain and also high nonspecific uptake in the pituitary (1.8 ± 0.3 ID at 30 min), a region with high β2-ARs levels. No further studies were reported using 26 to image β-ARs.
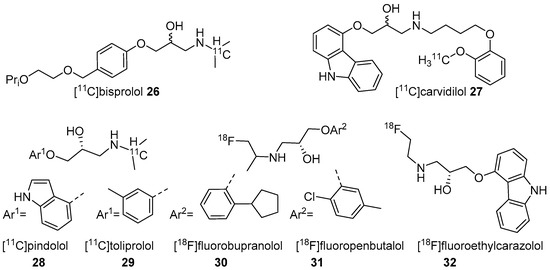
Figure 2. Radiotracers based on various β-AR blockers.
The five radiotracers had strong affinities (subnanomolar Kd) for β1 and β2-ARs. Although these radiotracers had sufficient affinity and lipophilicity for in vivo imaging, none showed good brain uptake. This group also evaluated S-[18F]fluoroethylcarazolol (32, β1 Ki = 0.5 nM, β2 Ki = 0.4 nM and Log P7.4 = 1.94) for in vivo imaging of β-ARs in rat brain [119]. The radiotracer 32 (Figure 2) was prepared via an epoxide ring-opening using [18F]fluoroethylamine and the corresponding epoxide with >10 GBq/μmol molar activity. The radiotracer accumulated in brain with uptake reflecting cerebral β-ARs binding. However, no further PET imaging studies were reported using 32 probably because of its analogous nature to 23 which was shown to be positive Ames test [114,120].
This entry is adapted from the peer-reviewed paper 10.3390/molecules25174017