TAM receptors (Tyro3, Axl and MerTK) are a family of tyrosine kinase receptors that are expressed in a variety of cell populations, including liver parenchymal and non-parenchymal cells These receptors are vital for immune homeostasis, as they regulate the innate immune response by suppressing inflammation via toll-like receptor inhibition and by promoting tissue resolution through efferocytosis. However, there is increasing evidence indicating that aberrant TAM receptor signaling may play a role in pathophysiological processes in the context of liver disease. This review will explore the roles of TAM receptors and their ligands in liver homeostasis as well as a variety of disease settings, including acute liver injury, steatosis, fibrosis, cirrhosis-associated immune dysfunction and hepatocellular carcinoma. A better understanding of our current knowledge of TAM receptors in liver disease may identify new opportunities for disease monitoring as well as novel therapeutic targets. Nonetheless, this review also aims to highlight areas where further research on TAM receptor biology in liver disease is required.
- TAM receptors
- Axl
- MerTK
- liver inflammation
- cirrhosis
1. Biology of TAM Receptors
1.1. TAM Receptor and Ligand Structure
TAM receptors are one of the 20 families of receptor tyrosine kinases. They comprise three receptors that share a similar structure: Tyro-3, Axl and MerTK. TAM receptors are expressed in a variety of tissues, and their expression patterns in the body have been reviewed extensively [1]. Briefly, Axl is expressed in cell populations in the liver, kidney, heart, skeletal muscle, testis and cerebellum but also in blood circulating cells such as monocytes and activated platelets [2]. In addition, Axl and its ligand Gas6 are expressed by endothelial cells [3]. Tyro3 expression is confined mostly to the central nervous system; it is expressed by a variety of tissues including the cerebral cortex, cerebellum, olfactory bulbs and amygdala, but also by platelets [2][4][5]. MerTK is expressed in cell populations in ovaries, testis, liver, lung, kidney, cerebral cortex and retina, but also in blood circulating cells, such as natural killer cells, platelets and monocytes [2][6]. The structure of TAM receptors consists of an extracellular domain (EC) defined by two tandem N-terminus immunoglobulin-like domains (Ig-like) (see Figure 1), which allow the interaction between receptor and ligand, as well as two fibronectin type III (FNIII) domains. The EC domain is followed by a transmembrane domain (TM) and finally, an intracellular C-terminus tyrosine kinase domain (TKD) [7]. The activation of TAM receptors depends on the binding of the EC domain by its ligands. Gas6 and Protein S (known as Pros1) are the two most well-known ligands and share a high degree of structural homology [8][9]. Although other ligands for TAM receptors have been identified, such as galectin-3 and Tubby-like protein 1 (Tulp-1), these have not been extensively studied and their physiologic roles remain relatively unknown [10][11].
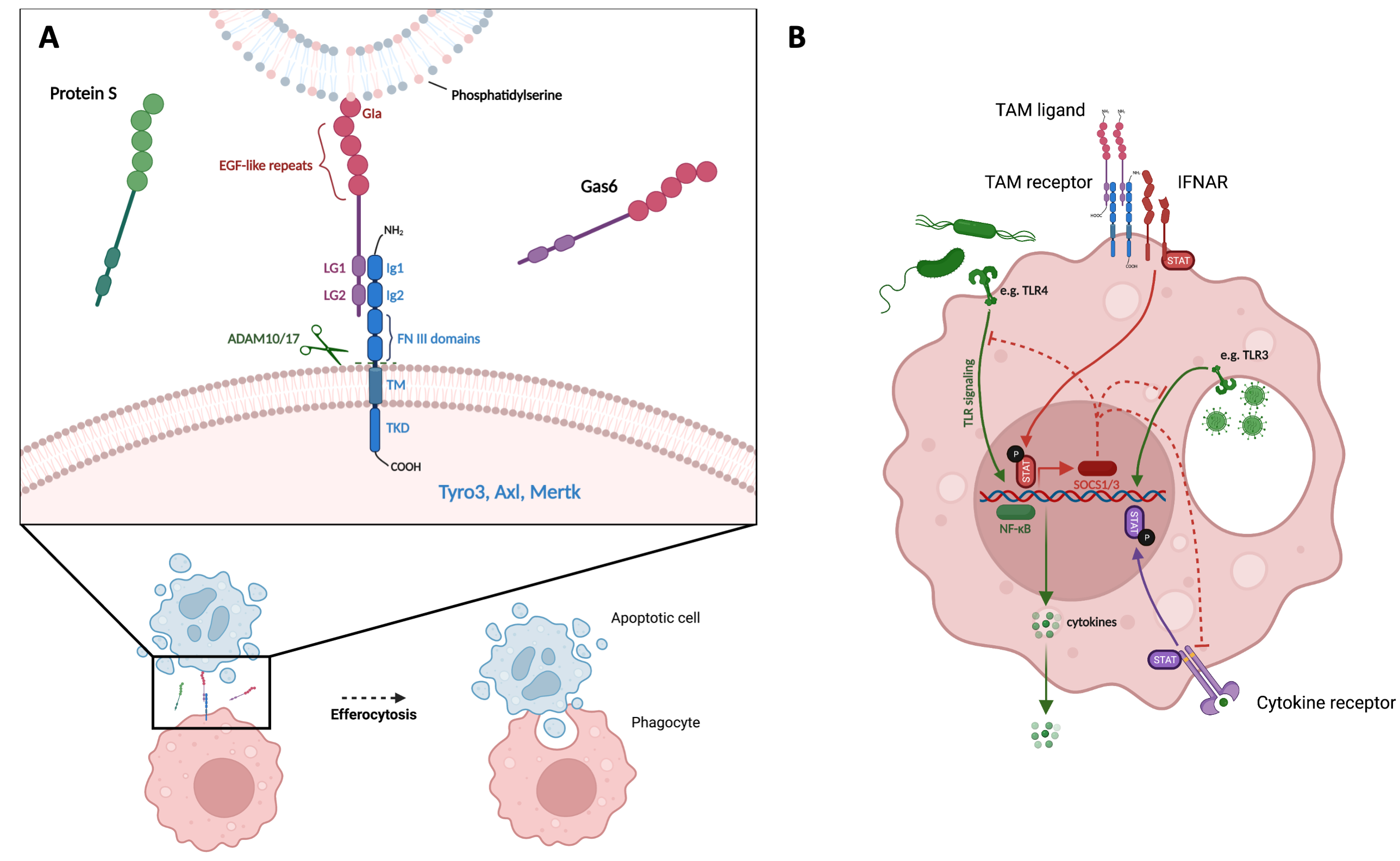
Figure 1. TAM receptors promote efferocytosis and inhibit TLR signaling. (A) TAM receptors promote efferocytosis. Illustration of TAM ligand (Gas6) serving as a bridge between PtdSer exposed on the surface of an apoptotic cell and a TAM receptor expressed by a phagocyte. Ligand G1a domain binds PtdSer on apoptotic cell; LG domains of ligand bind to Ig domains of TAM receptor; ADAM10/17 cleave TAM receptor at cleavage sites within TM domain. (B) TAM receptors inhibit TLR signaling. Illustration of TAM ligand binding and activation of TAM receptor, interaction with IFNAR/STAT complex, transcription of SOCS1/3, inhibition of TLR- and cytokine receptor-signaling pathways, and reduction in NF-κB-induced pro-inflammatory cytokine production. PtdSer = Phosphatidylserine, EGF = epidermal growth factor, LG = laminin G, Ig = immunoglobulin-like, FNIII = fibronectin type III, ADAM10/17 = A Disintegrin and metalloproteinase domain-containing protein 10/17, TM = transmembrane, TKD = tyrosine kinase domain, IFNAR = type I interferon receptor, TLR = toll-like receptor, SOCS1/3 = suppressors of cytokine signaling 1 and 3, STAT = signal transducer and activator of transcription, NF-κB = nuclear factor kappa-light-chain-enhancer of activated B cells.
Ligand structure consists of three main elements: the N-terminus has a γ-carboxyglutamic acid (Gla) domain that is followed by four epidermal growth factor (EGF)-like repeats. Adjacent to the C-terminus are two laminin G (LG) domains [12]. Gas6 and Protein S have different affinities for the three TAM receptors. Gas6 binds all receptors with the following order of affinity Axl > Tyro-3 > MerTK, while Protein S binds Tyro-3 and MerTK but not Axl [13]. The pattern of TAM receptor activation is defined by an initial binding of the ligand LG-like domains to the TAM receptor’s Ig-like domains, which leads to receptor dimerization, with subsequent trans-autophosphorylation of the kinase domains and finally, the activation of intracellular signaling cascades [14][15].
1.2. Function of TAM Receptors
TAM receptors play an important role in the regulation of the innate immune response. In particular, they act as a brake pedal for inflammatory responses and promote tissue resolution. Firstly, these receptors regulate the phagocytosis of apoptotic cells (namely efferocytosis), a fundamental process for the restoration of immune and tissue homeostasis. Mechanistically, phosphatidyl-serine (PtdSer) exposed on the membrane surface of apoptotic cells can bind to TAM receptors, thereby initiating uptake. Moreover, in the presence of Ca2+ ions, PtdSer binds the G1a domain of either Gas6 or Protein S [16], which increases the uptake of apoptotic bodies by macrophages as well as other phagocytes [14][15]. A study illustrating the vital role of TAM receptors in tissue homeostasis was conducted by generating triple TAM knockout (KO) mice, which were infertile by 3 weeks of age due to abnormal testis development. In fact, Sertoli cells express all three TAM receptors; accordingly, these mice displayed an accumulation of apoptotic spermatogenic cells [17]. Secondly, through suppression of Toll-like receptor (TLR) signaling, TAM receptors serve as a negative feedback loop for the innate immune response. In detail, following ligand-mediated auto-phosphorylation, these receptors interact with the type I interferon receptor (IFNAR)/STAT1 complex, which initially amplifies the inflammatory response; however, this association causes functional changes of IFNAR/STAT towards an anti-inflammatory molecule, which in turn induces transcription of the suppressors of cytokine signaling 1 and 3 (SOCS1 and 3) proteins. These proteins ultimately inhibit both TLR as well as cytokine receptor pro-inflammatory signaling [18]. Furthermore, TAM receptors may play a role in viral infection [19]; notably, these receptors may be implicated in virus–host interplay during SARS-CoV2 infection, which was recently reviewed in detail [20].
1.3. Regulation of TAM Receptor Expression
The regulation of the TAM receptor expression has been detailed both at transcriptional and post-transcriptional levels. Regarding the former, an increase in AXL gene expression was found in human monocyte-derived macrophages treated with the macrophage colony-stimulating factor, compared to cells treated with the granulocyte-macrophage colony-stimulating factor [21]. However, inflammatory stimuli, such as lipopolysaccharide (LPS), can also drive Axl expression [22], whereas, upregulation of MerTK expression has been shown for both pro- and anti-inflammatory cytokines, such as IL-17A and IL-10, respectively [23]. Additionally, glucocorticoids and Liver X receptor stimulation increase MerTK expression [24][25]. Tyro-3 remains the least well-studied member of the TAM receptor family and the regulation of its expression is currently not well established. Several studies on macrophages within the tumour microenvironment have characterized TAM receptor expression on both anti-tumour and pro-tumour macrophages. Of note, monocyte differentiation into macrophages upregulates MerTK expression, although the same studies observed contrasting results regarding changes in Axl expression upon macrophage differentiation [26][27]. Whilst high MerTK expression has been consistently attributed to an immunosuppressive, pro-restorative phenotype [28][29], upregulation of Axl has been observed in both macrophages stimulated by TLR ligands and interferon-γ [30] as well as in cells stimulated with IL-4 or IL-13 stimulation [31].
At the post-transcriptional level, ectodomain shedding of TAM receptors seems to play an important role in the regulation of these receptors. In fact, the TM domain is characterised by cleavage sites for a disintegrin and metalloproteinase domain-containing protein 10 and 17, also known as ADAM10 and ADAM17. The proteolytic processing of TAM receptors by these proteins leads to cleavage and release of a soluble component, termed sAxl, sMer and sTyro-3, depending on the receptor [32][33]. Interestingly, these molecules maintain the ability to bind TAM ligands, thereby dampening ligand-induced signaling [34]. In addition to ectodomain shedding, which is well established, a recent study suggests that Axl can be processed by α- and γ-secretases, leading to the formation of an intracellular domain that translocates into the nucleus [35].
2. TAM Receptors in Liver Disease
2.1. Liver Homeostasis
The liver is continuously exposed to gut-derived bacteria and microbial products from the portal circulation and thus faces the unique challenge of serving as an immunological barrier while also maintaining immune tolerance. One of the first studies investigating the role of TAM receptors in the liver revealed that these receptors are key for maintaining liver immune tolerance. In fact, triple KO mice (Axl−/−, Mertk−/−, Tyro3−/−) spontaneously developed inflammation, which persisted, leading to chronic inflammation [36]. Furthermore, investigations into the differential expression of TAM receptors reveal that Axl and MerTK are rarely co-expressed in mice [30]. Rather, bone marrow-derived macrophages and bone marrow-derived dendritic cells cultured in vitro expressed MerTK or Axl almost exclusively, respectively. These findings were confirmed in vivo in murine macrophages and dendritic cells of the spleen and lung. However, murine liver-resident macrophages are an exception to this rule and displayed co-expression of Axl and MerTK, although Tyro3 expression was absent in KC [30]. Moreover, MerTK, but not Axl, is known to be ubiquitous in murine tissue-resident macrophages, including liver-resident macrophages [37]. The role of TAM receptors in liver homeostasis was recently dissected by Zagórska et al. in Cx3cr1-specific double KO mice (Axl−/− Mertk−/−). In Axl- and MerTK-deficient mice, apoptotic cells accumulated in the liver with ageing. Furthermore, these mice displayed inflammatory infiltrates in the liver, paralleled by increased liver mRNA levels of pro-inflammatory cytokines and chemokines as well as serum levels of liver damage markers. Thus, in accordance with the known functions of TAM receptors, Axl and Mer promoted efferocytosis and prevented excessive immune activation in the liver [38]. However, further investigations into the role of TAM receptors in tolerogenic barrier immunity of the liver are needed.
2.2. Acute Liver Injury
MerTK seems to play a vital role in acute sterile injury, as displayed by a study in acute liver failure (ALF). Interestingly, a distinct subset of cells with high HLA-DR and MerTK expression was expanded in both circulation and tissue compartments of ALF patients. Notably, this subset was confirmed in an ALF model of acetaminophen (APAP)-treated mice and the appearance of these cells was restricted to the resolution phase of liver injury. The role of MerTK in liver repair and hepatoprotection is evident in APAP-treated Mertk−/− mice, which displayed persistent hallmarks of liver injury, including lower proportions of resident KCs and increased number of activated hepatic neutrophils during both peak and resolution phases. In ALF patients, MerTK + HLA-DRhigh monocytes displayed enhanced efferocytosis of apoptotic cells as well as clearance of Escherichia Coli. Thus, myeloid reprogramming towards this pro-restorative phenotype may be of therapeutic significance, as displayed by the beneficial effects of secretory leucocyte protease inhibitor (SLPI) both in vitro and in APAP-treated mice [39]. Of note, Axl−/− mice treated with APAP showed no changes in efferocytosis. Nonetheless, similarly to Mertk deficient mice, Axl−/− mice exhibited signs of increased liver damage, including massive haemorrhage and elevated serum levels of ALT in comparison to Mertk−/− and wild-type mice, indicating that Axl may play a role in alleviating APAP-induced liver injury [38]. In line with these findings, Gas6 signaling alleviated ischemia/reperfusion (I/R) injury in a mouse model of liver I/R. Moreover, in vitro experiments suggested that TAM receptor signaling protected hepatocytes from hypoxia-induced death and dampened cytokine expression of IL-1β and TNF by murine macrophages. In fact, Gas6 treatment improved liver I/R injury in both Gas6−/− mice and wild type mice, which highlights its potential use as a therapeutic in post-ischemic hepatic damage [40]. These studies on TAM signaling in acute liver injury validate previous findings, demonstrating that Gas6−/− mice exhibited a defective wound healing response following carbon tetrachloride (CCl4)-induced toxic hepatitis, accompanied by impaired KC activation [41].
2.3. Liver Steatosis
Several studies have shed light on Axl and MerTK signaling in steatosis, which seem to play different or even divergent roles in this context. Axl inhibition in a NASH model led to decreased cytokine production by LPS-stimulated KCs, as well as a reduction in expression of genes related to inflammation and fibrosis. Interestingly, Mertk−/− mice displayed an enhanced phenotype of NASH, whereas Axl−/− mice showed a partial improvement of disease hallmarks, such as decreased liver fibrosis and inflammation. Moreover, Gas6 stimulation of primary hepatocytes attenuated palmitic acid-induced lipotoxicity via activation of MerTK, while Axl activation showed no beneficial effects on lipotoxicity [42]. In contrast, a cross-sectional cohort study revealed that NAFLD patients who were carriers of a loss-of-function MERTK variant (rs4374383 G > A) displayed reduced activation of the pro-inflammatory NF-κB pathway in mononuclear cells upon oral fat tolerance test, as well as higher fat oxidation rates and increased tissue insulin sensitivity. In fact, the rs4374383 G > A variant protected healthy nonobese nondiabetic patients from 9-year incident NAFLD [43]. In addition, a study of the role of Gas6 in an experimental model of steatohepatitis revealed that Gas6 deficiency delays steatosis. Interestingly, these Gas6−/− mice also displayed restored expression levels of genes involved in β-oxidation, as well as increased expression of PPARα [44]. Finally, the cleavage of MerTK by ADAM17 is reduced in NASH, which leads to increased MerTK signaling in KC [45].
2.4. Liver Fibrosis
There is increasing evidence suggesting that sustained TAM signaling may play a role in liver fibrosis and thus could be detrimental in the context of chronic liver injury. The critical role of activated HSC as drivers of liver fibrosis is well established. Interestingly, the activation of Axl increased primary mouse HSC activation while Axl siRNA blocked the process. In vivo, both Axl knockout and Axl inhibition via bemcentinib (BGB324) led to reduced collagen deposition in a CCL4-induced liver fibrosis model [46]. Accordingly, it has been shown that Gas6 is secreted by activated HSC following injury in a rat CCL4-induced fibrosis model. Furthermore, Gas6 promotes activated HSC survival via a vitamin K-dependent anti-apoptotic effect and thus may contribute to activated HSC accumulation in liver fibrosis [47]. Moreover, Gas6 deficiency improved liver fibrosis in a steatohepatitis mouse model due to reduction in myofibroblast activation as shown by decreased expression of transforming growth factor beta (TGF-β) and collagen 1 mRNAs [44]. In addition to the role of Gas6/Axl signaling, there are studies indicating that MerTK signaling is also involved in liver fibrosis. Firstly, the progression of fibrosis in non-alcoholic fatty liver disease (NAFLD) patients is predicted by a specific MerTK rs4374383 genotype associated with increased hepatic MerTK expression [43]. Secondly, a MerTK variant associated with an allele-specific downregulation of MerTK following treatment of hepatitis C with interferon-α is associated with reduced risk of liver fibrosis in genome-wide association studies [48]. More recently, a molecular mechanism underlying these studies on MerTK variants as genetic risk factors was proposed. The targeting of MerTK specifically in myeloid cells in a mouse model of nonalcoholic steatohepatitis (NASH) reduced liver fibrosis. Accordingly, mice with a MerTK receptor that was cleavage-resistant to ADAM17 displayed increased liver fibrosis. Moreover, an increase in TGFβ1 following activation of MerTK on macrophages led to HSC activation with increased collagen production [45]. Interestingly, TAM receptors and their ligands, especially Protein S, regulate hemostasis (previously reviewed in detail [49]). Moreover, patients with liver cirrhosis display changes in hemostasis, leading to thrombotic or hemorrhagic events, which are associated with high morbidity and mortality [50]. Thus, a potential area of research, which remains currently underexplored, is the role of TAM receptors in cirrhosis-associated hemostatic alterations.
2.5. Cirrhosis-Associated Immune Dysfunction
A better understanding of not only the hallmarks of cirrhosis-associated immune dysfunction (CAID) but also bacterial translocation in cirrhosis is required to contextualize recent findings related to TAM receptors in liver disease. Firstly, although bacterial translocation occurs from the gut to mesenteric lymph nodes in healthy conditions and is vital for host immunity, this process is increased in liver cirrhosis and exceeds the host’s ability to maintain tolerance [51]. Secondly, the progression of cirrhosis is accompanied by systemic inflammation, as shown by increased serum pro-inflammatory cytokines [52], as well as the activation of circulating immune cells that display changes both in phenotype and function [53][54][55][56]. In parallel, dysfunctional hepatic protein synthesis leads to an impaired complement system and reduced pathogen recognition [57][58], along with reduced KC numbers [59] and an expansion of immunosuppressive circulating immune cells [22][60]. These mechanisms together have been proposed to underlie the increased susceptibility to infections (e.g., spontaneous bacterial peritonitis) and relate to mortality in patients with cirrhosis [61][62][63].
Several studies suggest that TAM receptor signaling plays a role in CAID. It was shown that Gas6/Axl signaling in liver macrophages induced autophagy, which in turn prevented NLRP3 inflammasone activation and inhibited the hepatic inflammatory response. Moreover, Axl−/− mice were partially protected from hepatic injury induced by either LPS or CCL4 compared to wild-type mice [64]. As previously mentioned, MerTK-expressing immunoregulatory monocytes were expanded in the circulation of patients with acute-on-chronic liver failure (ACLF) compared to not only healthy controls but also patients with stable cirrhosis. In parallel, MerTK-expressing macrophages were increased in the liver, peritoneum and mesenteric lymph nodes of these patients. Remarkably, MerTK expression was correlated with hepatic (Child-Pugh and MELD), as well as extrahepatic (CLIF-SOFA and NACSELD) disease severity scores as well as with the systemic inflammatory response (SIRS). The response of MerTK-expressing monocytes ex vivo to microbial exposure indicated that these cells show attenuated production of pro-inflammatory cytokines IL-6 and TNF-α. Accordingly, MerTK+ monocytes displayed an anti-inflammatory phenotype. Moreover, in vitro experiments suggested that MerTK-expressing cells displayed increased migration into inflamed tissues in the context of cirrhosis-associated endothelial dysfunction, in fact, repeated cycles of migration may constitute a mechanism for MerTK upregulation in ACLF [60]. In addition to these findings on MerTK in ACLF, Axl-expressing monocytes were expanded in the circulation of patients along early and advanced stages of cirrhosis in the absence of acute decompensation. These cells produced lower levels of inflammatory cytokines IL-6 and TNF-α in response to LPS stimulation and displayed reduced T-cell activation and increased phagocytic removal of apoptotic cells. AXL-expression correlated not only with disease severity scores, such as Child-Pugh, MELD and classification by D’Amico, but also with major complications of cirrhosis, such as ascites, varices and hepatic encephalopathy. Moreover, high Axl-expression predicted development of infection, onset of acute decompensation and mortality in patients with cirrhosis. Mechanistically, Axl is upregulated upon exposure to pathogen-associated molecular patterns (PAMPs), as well as uptake of either apoptotic cells or bacteria. Importantly, Axl inhibition restored inflammatory cytokine production by monocytes ex-vivo, which highlights a potential therapeutic target in CAID [22].
Several studies suggest that plasma components of the TAM system may also serve as noninvasive markers of chronic liver disease complications. In particular, plasma Gas6 may be a promising biomarker for the detection of oesophageal varices and may represent an alternative to Baveno VI criteria in those cases where transient elastography (TE) is unavailable or unsuccessful due to cost constraints or patient obesity, respectively [65]. Moreover, plasma sAxl levels were an accurate biomarker for liver fibrosis and cirrhosis in patients with NAFLD or viral hepatitis; additionally, the accuracy can be increased by incorporating albumin into a sAxl/albumin ratio, which represents another suitable alternative to TE [66]. In fact, plasma Gas6 levels have been shown to be elevated in patients with cirrhosis across multiple studies, as well as increasing in parallel with severity of cirrhosis. Moreover, sAxl, sMer and Protein S were increased in patients with cirrhosis, while Galectin-3 was specifically elevated in patients with ACLF [22][42][60].
2.6. Hepatocellular Carcinoma
While TAM receptors have been investigated in a wide variety of cancers, including non-small-cell lung cancer, acute myeloid leukemia, chronic myeloid leukemia, breast cancer, ovarian cancer, glioblastoma, and melanoma [67], findings related to hepatocellular carcinoma (HCC) are currently limited and are restricted to the role of Axl. Of note, not only liver non-parenchymal cells but also malignant hepatocytes express Axl in HCC [68]. Although TAM receptor signaling may be considered anti-oncogenic due to its role in preventing chronic inflammation, especially at early stages of disease, studies on Axl signaling in HCC have revealed a multitude of possible oncogenic roles as well. Firstly, as previously described, both sustained Axl and sustained MerTK signaling in non-parenchymal liver cells may drive liver fibrosis, which may predispose these patients to develop HCC. Secondly, in vitro experiments indicate that Axl signaling may confer the invasive and migratory characteristics of hepatocytes that have undergone epithelial-to-mesenchymal transition (EMT). Accordingly, HCC patients with high Axl expression also displayed increased vessel invasion of HCC cells, increased risk of tumour recurrence following liver transplantation and drastically lower survival rates [69]. Finally, since it is known that TAM receptor signaling in macrophages of the tumour microenvironment polarizes these cells towards an immunosuppressive state (also known as pro-tumour macrophages) [70], one may speculate that this mechanism occurs in HCC as well. However, evidence for this process is currently lacking in HCC. In addition to these oncogenic mechanisms, it was shown that a combination of transforming growth factor-ß signaling and Axl signaling lead to CXCL5 expression and release, which is known to drive HCC progression via recruitment and infiltration of neutrophils into tumour tissue. Accordingly, both CRISPR/Cas9-mediated Axl knockout and gene silencing of Axl abrogated CXCL5 levels in HCC cell lines, which reduced the chemokine-dependent attraction of neutrophils into HCC tumours [71]. A recent study revealed the role of Axl overexpression in portal vein tumour thrombus (PVTT), which was associated with poor overall survival (OS). AXL overexpression in tumour-derived endothelial cells, but not tumour cells, was associated with reduced OS in HCC patients with PVTT. Furthermore, high AXL expression promoted HCC cell migration and metastasis in both in vitro and in vivo xenograft models. Accordingly, the increase in metastasis was abolished upon treatment with an Axl inhibitor (R428) [72]. Another study related to HCC metastasis detailed an important mechanism of AXL gene regulation. The splicing regulator PTBP1 led to the formation of a spliced variant of Axl with increased binding affinity to Gas6 and downstream signaling, which drove liver cancer cell migration, invasion and metastasis in both in vitro but also in vivo settings [73]. Finally, a study investigated expression levels of the tumour suppressor miRNA-34a-5p in HCC tissues, which indicated that high expression was associated with better survival. Importantly, miRNA-34a-5p targets AXL and transfection in an HCC cell line led to reduced proliferation and increased apoptosis as well as decreased chemoresistance to cisplatin. In fact, these beneficial effects can be recapitulated upon transfection with small interfering RNA for AXL, which highlights the multifaceted role of Axl in HCC [74].
In addition to studies investigating the role of Axl in HCC, the diagnostic utility of blood sAxl levels in HCC patients has been explored. In fact, a study indicated that sAxl might outperform the well-established serum marker Alpha-foetoprotein (AFP) in detecting HCC. In particular, sAxl was superior in early HCC and in AFP-negative HCC. Moreover, by combining sAxl and AFP, the specificity for early HCC diagnosis was improved [75] [76]. In support of sAxl as a specific marker for cirrhosis and HCC, a large-scale multicentre analysis indicated that serum levels remain unvaried in many other chronic liver diseases, such as cholestatic liver disease, chronic viral hepatitis and autoimmune hepatitis, as well as liver adenomas and cholangiocarcinomas (CCA) [77]. Interestingly, the multi-kinase inhibitor Cabozantinib is already approved for use in patients with sorafenib-pretreated advanced HCC. Of note, this inhibitor does not impact Axl alone, but in fact it displays higher selectivity for the vascular endothelial growth factor (VEGF) receptor 2 and mesenchymal-epithelial transition factor (MET) receptor. Thus, it is hard to discern the specific role of Axl in studies involving Cabozantinib treatment in HCC [78]. This scenario is complicated further by supposing that Axl modulation may impact tumour cells indirectly via Axl-expressing macrophages in the tumour microenvironment [70].
2.7. Cholestatic Diseases and Cholangiocarcinoma
Lastly, a small number of studies highlighted potential roles of Axl in both cholestatic disease and CCA. High serum Axl and Gas6 levels have been proposed as non-invasive biomarkers for advanced histological stage in patients with primary biliary cholangitis. Moreover, the diagnostic accuracy of these markers was further improved by incorporating albumin into Axl/albumin and Gas6/albumin ratios [79]. In the context of CCA, AXL/signal transducer and activator of the transcription 3 (AXL/STAT3) inactivation by the opioid-binding protein/cell adhesion molecule, such as (OPCML), a well-known tumour suppressor, induced apoptosis and inhibited cell proliferation in CCA cell lines [80]. In addition, a novel histone deacetylase inhibitor (CG200745) displaying anticancer effects in CCA both in vitro and in vivo, downregulated Axl and Gas6 genes in two CCA cell lines [81]. Finally, metformin reduced human CCA cell proliferation both in vitro and in vivo, which was associated with decreased phosphorylation of a variety of tyrosine kinase receptors, including Axl [82].
3. Conclusions and Future Questions
Axl and MerTK seem to play an important role in liver immune homeostasis, by suppressing inflammatory responses and promoting the removal of apoptotic cells. Accordingly, TAM receptor-deficient mice develop chronic liver inflammation. Furthermore, myeloid-specific deletion of Axl and MerTK leads to liver damage, apoptotic cell accumulation and inflammatory infiltrates in the livers of ageing mice. TAM receptors display differential expression in myeloid cell subsets across liver and extrahepatic compartments (see Table 1). In addition, in the context of liver disease these two receptors can have diverse roles depending on the liver pathophysiology (see Table 2). In summary, MerTK signaling is hepatoprotective and vital for tissue resolution following acute liver injury. In liver steatosis, evidence in support of the role of TAM receptors is currently sparse. Briefly, Axl signaling exacerbated inflammation and fibrosis in an experimental model of NASH, whereas MerTK signaling seemed to attenuate these processes. Undoubtedly, sustained pathological Axl and MerTK signaling in non-parenchymal cells play a role in hepatic fibrosis. Although several mechanisms have been proposed, more detailed molecular mechanisms underlying TAM receptor-mediated liver fibrosis are needed. Moreover, Axl- and MerTK-expressing immunosuppressive monocytes are expanded in the circulation of patients with cirrhosis or acute-on-chronic liver failure, respectively. These findings add to the spectrum of immune alterations, known as CAID, and may provide a link with the increased susceptibility to infections and mortality in these patients. In HCC, Axl signaling in malignant parenchymal cells promotes survival, invasion, migration, EMT and chemoresistance, as well as neutrophil infiltration into the tumour tissue. Moreover, Axl overexpression in tumour-derived endothelial cells drives metastasis and is associated with poor survival in HCC patients with PVTT. In addition to the mechanisms of TAM receptor biology discussed here, it would seem that levels of circulating TAM system components, such as soluble receptors (sAxl) and ligands (Gas6 and Galectin-3), can serve as non-invasive biomarkers in a variety of pathologies including fibrosis, cirrhosis, chronic liver disease complications and HCC.
| Axl | MerTK | Tyro-3 | |
|---|---|---|---|
| Bone Marrow | 
|

|

|
| Blood | 

|


|

|
| Liver | 

|


|

|
 = murine data;
= murine data;  = human data. (Health vs. Disease).
= human data. (Health vs. Disease).| Liver Pathophysiology | Axl Signaling | MerTK Signaling |
|---|---|---|
| Homeostasis | 
|
|
| Acute liver injury | 
|

|
| Steatosis | 
|

|
| Fibrosis | 

|
|
| Cirrhosis-associated immune dysfunction | 
|

|
| Hepatocellular carcinoma | 
|
ø |
 = murine data;
= murine data;  = human data; ø = lack of findings.
= human data; ø = lack of findings.Given our current understanding of TAM receptor involvement in liver disease, one may speculate on different therapeutic approaches. In acute liver failure, the expansion of prorestorative hepatic MerTK+ macrophages by SLPI may promote tissue resolution and provide a therapeutic benefit. MerTK agonism may also constitute a promising strategy for the treatment of ALF; however, MerTK-specificity may be important, as a molecule displaying concurrent Axl agonism may have a pro-fibrotic effect on the liver. Moreover, since patients with NAFLD display an increase of Axl signaling during progression of disease and the Bemcentinib administration reduced liver fibrosis and inflammation in experimental NASH, Axl targeting, either directly or possibly indirectly via increased ectodomain cleavage by ADAM10/17, may be a therapeutic strategy for the treatment of patients with NASH. While MerTK targeting in a murine model of NASH also reduced liver fibrosis, MerTK seems to confer hepatocyte protection against lipotoxicity via Gas6, and thus it is not clear whether MerTK targeting may be of therapeutic benefit in the context of NASH. In patients with cirrhosis, Axl inhibition may counteract the immunosuppressive features of Axl-expressing monocytes in the circulation and prevent infections, which constitute the leading cause of decompensation in cirrhosis. Analogously, MerTK inhibition in patients with ACLF may restore monocyte responses to bacterial challenge and prevent secondary infections. When evaluating TAM receptor modulation as a therapeutic approach, one must also consider unwanted systemic effects, since these receptors are ubiquitously expressed in the body and are vital for immune homeostasis. Currently, there are no approved therapies involving TAM receptor modulation in liver disease, except for Cabozantinib, which is not a selective Axl inhibitor per se. Given the compartmental regulation of TAM receptors on myeloid cells, in vivo studies are required to test and explore both the effects and side effects of TAM modulation in liver disease. However, due to the increasing number of preclinical trials evaluating TAM-targeting agents for the treatment of cancer, the idea of repurposing such agents for the treatment of liver diseases seems promising. In conclusion, there is a growing body of research related to distinct TAM receptor signaling in liver disease, highlighting the need for further research in this field.
This entry is adapted from the peer-reviewed paper 10.3390/livers2010002
References
- Linger, R.M.; Keating, A.K.; Earp, H.S.; Graham, D.K. TAM Receptor Tyrosine Kinases: Biologic Functions, Signaling, and Potential Therapeutic Targeting in Human Cancer. Adv. Cancer Res. 2008, 100, 35–83.
- Angelillo-Scherrer, A.; Burnier, L.; Flores, N.; Savi, P.; DeMol, M.; Schaeffer, P.; Herbert, J.; Lemke, G.; Goff, S.P.; Matsushima, G.K.; et al. Role of Gas6 receptors in platelet signaling during thrombus stabilization and implications for antithrombotic therapy. J. Clin. Investig. 2005, 115, 237–246.
- Holland, S.J.; Powell, M.J.; Franci, C.; Chan, E.W.; Friera, A.M.; Atchison, R.E.; McLaughlin, J.; Swift, S.E.; Pali, E.S.; Yam, G.; et al. Multiple roles for the receptor tyrosine kinase Axl in tumor formation. Cancer Res. 2005, 65, 9294–9303.
- Prieto, A.L.; Weber, J.L.; Lai, C. Expression of the Receptor Protein-Tyrosine Kinases Tyro-3, Axl, and Mer in the Developing Rat Central Nervous System. J. Comp. Neurol. 2000, 425, 295–314.
- Prieto, A.L.; Weber, J.L.; Tracy, S.; Heeb, M.J.; Lai, C. Gas6, a ligand for the receptor protein-tyrosine kinase Tyro-3, is widelyexpressed in the central nervous system. Brain Res. 1999, 816, 646–661.
- Pierce, A.M.; Keating, A.K. TAM receptor tyrosine kinases: Expression, disease and oncogenesis in the central nervous system. Brain Res. 2014, 1542, 206–220.
- Lemke, G. Biology of the TAM receptors. Cold Spring Harb. Perspect. Biol. 2013, 5, a009076.
- Manfioletti, G.; Brancolini, C.; Avanzi, G.; Schneider’, C. The Protein Encoded by a Growth Arrest-Specific Gene (gas6) Is a New Member of the Vitamin K-Dependent Proteins Related to Protein S, a Negative Coregulator in the Blood Coagulation Cascade. Mol. Cell. Biol. 1993, 13, 4976–4985.
- Dahlbäck, B.; Villoutreix, B.O. Regulation of blood coagulation by the protein C anticoagulant pathway: Novel insights into structure-function relationships and molecular recognition. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 1311–1320.
- Nomura, K.; Vilalta, A.; Allendorf, D.H.; Hornik, T.C.; Brown, G.C. Activated Microglia Desialylate and Phagocytose Cells via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J. Immunol. 2017, 198, 4792–4801.
- Caberoy, N.B.; Zhou, Y.; Li, W. Tubby and tubby-like protein 1 are new MerTK ligands for phagocytosis. EMBO J. 2010, 29, 3898–3910.
- Sasaki, T.; Knyazev, P.G.; Clout, N.J.; Cheburkin, Y.; Göhring, W.; Ullrich, A.; Timpl, R.; Hohenester, E. Structural basis for Gas6-Axl signalling. EMBO J. 2006, 25, 80–87.
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the Product of Growth Arrest-specific Gene 6 as a Common Ligand for Axl, Sky, and Mer Receptor Tyrosine Kinases. Cell Biol. Metab. 1996, 271, 30022–30027.
- Heiring, C.; Dahlbäck, B.; Muller, Y.A. Ligand recognition and homophilic interactions in Tyro3: Structural insights into the Axl/Tyro3 receptor tyrosine kinase family. J. Biol. Chem. 2004, 279, 6952–6958.
- Tsou, W.-I.; Nguyen, K.-Q.N.; Calarese, D.A.; Garforth, S.J.; Antes, A.L.; Smirnov, S.V.; Almo, S.C.; Birge, R.B.; Kotenko, S.V. Receptor Tyrosine Kinases, TYRO3, AXL, and MER, Demonstrate Distinct Patterns and Complex Regulation of Ligand-induced Activation. J. Biol. Chem. 2014, 289, 25750–25763.
- Graham, D.K.; DeRyckere, D.; Davies, K.D.; Earp, H.S. The TAM family: Phosphatidylserine-sensing receptor tyrosine kinases gone awry in cancer. Nat. Rev. Cancer 2014, 14, 769–785.
- Wang, H.; Chen, Y.; Ge, Y.; Ma, P.; Ma, Q.; Ma, J.; Wang, H.; Xue, S.; Han, D. Immunoexpression of Tyro 3 Family Receptors—Tyro 3, Axl, and Mer—and Their Ligand Gas6 in Postnatal Developing Mouse Testis. J. Histochem. Cytochem. 2005, 53, 1355–1364.
- Rothlin, C.V.; Ghosh, S.; Zuniga, E.I.; Oldstone, M.B.; Lemke, G. TAM Receptors Are Pleiotropic Inhibitors of the Innate Immune Response. Cell 2007, 131, 1124–1136.
- Wang, Z.-Y.; Wang, P.-G.; An, J. The Multifaceted Roles of TAM Receptors during Viral Infection. Virol. Sin. 2020, 36, 1–12.
- Tutusaus, A.; Marí, M.; Ortiz-Pérez, J.T.; Nicolaes, G.A.F.; Morales, A.; De Frutos, P.G. Role of Vitamin K-dependent Factors Protein S and GAS6 and TAM Receptors in SARS-CoV-2 Infection and COVID-19-Associated Immunothrombosis. Cells 2020, 9, 2186.
- Waterborg, C.E.J.; A Broeren, M.G.; Davidson, E.N.B.; I Koenders, M.; Lent, P.L.E.M.V.; Berg, W.B.V.D.; Van Der Kraan, P.M.; Loo, F.A.J.V.D. The level of synovial AXL expression determines the outcome of inflammatory arthritis, possibly depending on the upstream role of TGF-β1. Rheumatology 2018, 58, 536–546.
- Brenig, R.; Pop, O.T.; Triantafyllou, E.; Geng, A.; Singanayagam, A.; Perez-Shibayama, C.; Besse, L.; Cupovic, J.; Künzler, P.; Boldanova, T.; et al. Expression of AXL receptor tyrosine kinase relates to monocyte dysfunction and severity of cirrhosis. Life Sci. Alliance 2019, 3, e201900465.
- Zizzo, G.; Cohen, P.L. IL-17 Stimulates Differentiation of Human Anti-Inflammatory Macrophages and Phagocytosis of Apoptotic Neutrophils in Response to IL-10 and Glucocorticoids. J. Immunol. 2013, 190, 5237–5246.
- McColl, A.; Bournazos, S.; Franz, S.; Perretti, M.; Morgan, B.P.; Haslett, C.; Dransfield, I. Glucocorticoids Induce Protein S-Dependent Phagocytosis of Apoptotic Neutrophils by Human Macrophages. J. Immunol. 2009, 183, 2167–2175.
- Gonzalez, N.A.; Bensinger, S.J.; Hong, C.; Beceiro, S.; Bradley, M.N.; Zelcer, N.; Deniz, J.; Ramírez, C.; Díaz, M.; Gallardo, G.; et al. Apoptotic Cells Promote Their Own Clearance and Immune Tolerance through Activation of the Nuclear Receptor LXR. Immunity 2009, 31, 245–258.
- Zahuczky, G.; Kristóf, E.; Majai, G.; Fésüs, L. Differentiation and Glucocorticoid Regulated Apopto-Phagocytic Gene Expression Patterns in Human Macrophages. Role of Mertk in Enhanced Phagocytosis. PLoS ONE 2011, 6, e21349.
- Malawista, A.; Wang, X.; Trentalange, M.; Allore, H.G.; Montgomery, R.R. Coordinated expression of tyro3, axl, and mer receptors in macrophage ontogeny. Macrophage 2016, 3, e1261.
- Sanjurjo, L.; Aran, G.; Téllez, E.; Amézaga, N.; Armengol, C.; López, D.; Prats, C.; Sarrias, M.-R. CD5L Promotes M2 Macrophage Polarization through Autophagy-Mediated Upregulation of ID3. Front. Immunol. 2018, 9, 7.
- Grabiec, A.M.; Goenka, A.; E Fife, M.; Fujimori, T.; Hussell, T. Axl and MerTK receptor tyrosine kinases maintain human macrophage efferocytic capacity in the presence of viral triggers. Eur. J. Immunol. 2018, 48, 855–860.
- Zagórska, A.; Través, P.G.; Lew, E.D.; Dransfield, I.; Lemke, G. Diversification of TAM receptor tyrosine kinase function. Nat. Immunol. 2014, 15, 920–928.
- Shibata, T.; Habiel, D.M.; Coelho, A.L.; Kunkel, S.L.; Lukacs, N.W.; Hogaboam, C.M. Axl receptor blockade ameliorates pulmonary pathology resulting from primary viral infection and viral exacerbation of asthma. J. Immunol. 2014, 192, 3569–3581.
- Orme, J.; DU, Y.; Vanarsa, K.; Mayeux, J.; Li, L.; Mutwally, A.; Arriens, C.; Min, S.; Hutcheson, J.; Davis, L.S.; et al. Heightened cleavage of Axl receptor tyrosine kinase by ADAM metalloproteases may contribute to disease pathogenesis in SLE. Clin. Immunol. 2016, 169, 58–68.
- O’Bryan, J.P.; Fridell, Y.; Koski, R.; Varnum, B.; Liu, E.T. The Transforming Receptor Tyrosine Kinase, Axl, Is Post-translationally Regulated by Proteolytic Cleavage. J. Biol. Chem. 1995, 270, 551–557.
- Ekman, C.; Stenhoff, J.; Dahlbäck, B. Gas6 is complexed to the soluble tyrosine kinase receptor Axl in human blood. J. Thromb. Haemost. 2010, 8, 838–844.
- Lu, Y.; Wan, J.; Yang, Z.; Lei, X.; Niu, Q.; Jiang, L.; Passtoors, W.M.; Zang, A.; Fraering, P.C.; Wu, F. Regulated intramembrane proteolysis of the AXL receptor kinase generates an intracellular domain that localizes in the nucleus of cancer cells. FASEB J. 2016, 31, 1382–1397.
- Qi, N.; Liu, P.; Zhang, Y.; Wu, H.; Chen, Y.; Han, D. Development of a Spontaneous Liver Disease Resembling Autoimmune Hepatitis in Mice Lacking Tyro3, Axl and Mer Receptor Tyrosine Kinases. PLoS ONE 2013, 8, e66604.
- Gautier, E.L.; Shay, T.; Miller, J.; Greter, M.; Jakubzick, C.; Ivanov, S.; Helft, J.; Chow, A.; Elpek, K.G.; Gordonov, S.; et al. Gene-expression profiles and transcriptional regulatory pathways that underlie the identity and diversity of mouse tissue macrophages. Nat. Immunol. 2012, 13, 1118–1128.
- Zagórska, A.; Través, P.G.; Jiménez-García, L.; Strickland, J.D.; Oh, J.; Tapia, F.J.; Mayoral, R.; Burrola, P.; Copple, B.L.; Lemke, G. Differential regulation of hepatic physiology and injury by the TAM receptors Axl and Mer. Life Sci. Alliance 2020, 3, e202000694.
- Triantafyllou, E.; Pop, O.T.; Possamai, L.A.; Wilhelm, A.; Liaskou, E.; Singanayagam, A.; Bernsmeier, C.; Khamri, W.; Petts, G.; Dargue, R.; et al. MerTK expressing hepatic macrophages promote the resolution of inflammation in acute liver failure. Gut 2018, 67, 333–347.
- Llacuna, L.; Bárcena, C.; Bellido-Martín, L.; Fernández, L.; Stefanovic, M.; Marí, M.; García-Ruiz, C.; Fernández-Checa, J.C.; de Frutos, P.G.; Morales, A. Growth arrest-specific protein 6 is hepatoprotective against murine ischemia/reperfusion injury. Hepatology 2010, 52, 1371–1379.
- Lafdil, F.; Chobert, M.-N.; Deveaux, V.; Zafrani, E.-S.; Mavier, P.; Nakano, T.; Laperche, Y.; Brouillet, A. Growth arrest-specific protein 6 deficiency impairs liver tissue repair after acute toxic hepatitis in mice. J. Hepatol. 2009, 51, 55–66.
- Tutusaus, A.; de Gregorio, E.; Cucarull, B.; Cristóbal, H.; Aresté, C.; Graupera, I.; Coll, M.; Colell, A.; Gausdal, G.; Lorens, J.B.; et al. A Functional Role of GAS6/TAM in Nonalcoholic Steatohepatitis Progression Implicates AXL as Therapeutic Target. Cell. Mol. Gastroenterol. Hepatol. 2020, 9, 349–368.
- Petta, S.; Valenti, L.; Marra, F.; Grimaudo, S.; Tripodo, C.; Bugianesi, E.; Cammà, C.; Cappon, A.; Di Marco, V.; Di Maira, G.; et al. MERTK rs4374383 polymorphism affects the severity of fibrosis in non-alcoholic fatty liver disease. J. Hepatol. 2016, 64, 682–690.
- Fourcot, A.; Couchie, D.; Chobert, M.; Zafrani, E.; Mavier, P.; Laperche, Y.; Brouillet, A. Gas6 deficiency prevents liver inflammation, steatohepatitis, and fibrosis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2011, 300, 1043–1053.
- Cai, B.; Dongiovanni, P.; Corey, K.E.; Wang, X.; Shmarakov, I.O.; Zheng, Z.; Kasikara, C.; Davra, V.; Meroni, M.; Chung, R.T.; et al. Macrophage MerTK Promotes Liver Fibrosis in Nonalcoholic Steatohepatitis. Cell Metab. 2019, 31, 406–421.e7.
- Bárcena, C.; Stefanovic, M.; Tutusaus, A.; Joannas, L.; Menéndez, A.; García-Ruiz, C.; Sancho-Bru, P.; Caballería, J.; Rothlin, C.V. Gas6/Axl pathway is activated in chronic liver disease and its targeting reduces fibrosis via hepatic stellate cell inactivation. J. Hepatol. 2015, 63, 670–678.
- Lafdil, F.; Chobert, M.N.; Couchie, D.; Brouillet, A.; Zafrani, E.S.; Mavier, P.; Laperche, Y. Induction of Gas6 protein in CCl4-induced rat liver injury and anti-apoptotic effect on hepatic stellate cells. Hepatology 2006, 44, 228–239.
- Cavalli, M.; Pan, G.; Nord, H.; Arzt, E.W.; Wallerman, O.; Wadelius, C. Genetic prevention of hepatitis C virus-induced liver fibrosis by allele-specific downregulation of MERTK. Hepatol. Res. 2016, 47, 826–830.
- Van der Meer, J.H.M.; van der Poll, T.; van ’t Veer, C.V. TAM receptors, Gas6, and protein S: Roles in inflammation and hemostasis. Blood 2014, 123, 2460–2469.
- Zermatten, M.G.; Fraga, M.; Moradpour, D.; Calderara, D.B.; Aliotta, A.; Stirnimann, G.; De Gottardi, A.; Alberio, L. Hemostatic Alterations in Patients with Cirrhosis: From Primary Hemostasis to Fibrinolysis. Hepatology 2020, 71, 2135–2148.
- Wiest, R.; Lawson, M.; Geuking, M. Pathological bacterial translocation in liver cirrhosis. J. Hepatol. 2014, 60, 197–209.
- Albillos, A.; de la Hera, A.; Reyes, E.; Monserrat, J.; Muñoz, L.; Nieto, M.; Prieto, A.; Sanz, E.; Alvarez-Mon, M. Tumour necrosis factor-alpha expression by activated monocytes and altered T-cell homeostasis in ascitic alcoholic cirrhosis: Amelioration with norfloxacin. J. Hepatol. 2004, 40, 624–631.
- Muñoz, L.; Albillos, A.; Nieto, M.; Reyes, E.; Lledó, L.; Monserrat, J.; Sanz, E.; de la Hera, A.; Alvarez-Mon, M. Mesenteric Th1 polarization and monocyte TNF-α production: First steps to systemic inflammation in rats with cirrhosis. Hepatology 2005, 42, 411–419.
- Fiuza, C.; Salcedo, M.; Clemente, G.; Tellado, J.M. Granulocyte Colony-Stimulating Factor Improves Deficient In Vitro Neutrophil Transendothelial Migration in Patients with Advanced Liver Disease. Clin. Vaccine Immunol. 2002, 9, 433–439.
- Devière, J.; Content, J.; Denys, C.; Vandenbussche, P.; Schandene, L.; Wybran, J.; Dupont, E. Excessive In Vitro Bacterial Lipopolysaccharide-induced Production of Monokines in Cirrhosis. Hepatology 1990, 11, 628–634.
- Singanayagam, A.; Triantafyllou, E. Macrophages in Chronic Liver Failure: Diversity, Plasticity and Therapeutic Targeting. Front. Immunol. 2021, 12, 4.
- Runyon, B.A.; Morrissey, R.L.; Hoefs, J.C.; Wyle, F.A. Opsonic activity of human ascitic fluid: A potentially important protective mechanism against spontaneous bacterial peritonitis. Hepatology 1985, 5, 634–637.
- Helmy, K.Y.; Katschke, K.J., Jr.; Gorgani, N.N.; Kljavin, N.M.; Elliott, J.M.; Diehl, L.; Scales, S.J.; Ghilardi, N.; van Lookeren Campagne, M. CRIg: A Macrophage Complement Receptor Required for Phagocytosis of Circulating Pathogens. Cell 2006, 124, 915–927.
- Jenne, C.N.; Kubes, P. Immune surveillance by the liver. Nat. Immunol. 2013, 14, 996–1006.
- Bernsmeier, C.; Pop, O.T.; Singanayagam, A.; Triantafyllou, E.; Patel, V.; Weston, C.J.; Curbishley, S.; Sadiq, F.; Vergis, N.; Khamri, W.; et al. Patients With Acute-on-Chronic Liver Failure Have Increased Numbers of Regulatory Immune Cells Expressing the Receptor Tyrosine Kinase MERTK. Gastroenterology 2015, 148, 603–615.e14.
- Moreau, R.; Jalan, R.; Gines, P.; Pavesi, M.; Angeli, P.; Cordoba, J.; Durand, F.; Gustot, T.; Saliba, F.; Domenicali, M.; et al. Acute-on-Chronic Liver Failure Is a Distinct Syndrome That Develops in Patients with Acute Decompensation of Cirrhosis. Gastroenterology 2013, 144, 1426–1437.e9.
- Arvaniti, V.; D’Amico, G.; Fede, G.; Manousou, P.; Tsochatzis, E.; Pleguezuelo, M.; Burroughs, A.K. Infections in Patients with Cirrhosis Increase Mortality Four-Fold and Should Be Used in Determining Prognosis. Gastroenterology 2010, 139, 1246–1256.e5.
- Triantafyllou, E.; Woollard, K.J.; McPhail, M.; Antoniades, C.G.; Possamai, L.A. The Role of Monocytes and Macrophages in Acute and Acute-on-Chronic Liver Failure. Front. Immunol. 2018, 9, 2948.
- Han, J.; Bae, J.; Choi, C.-Y.; Choi, S.-P.; Kang, H.-S.; Jo, E.-K.; Park, J.; Lee, Y.S.; Moon, H.-S.; Park, C.-G.; et al. Autophagy induced by AXL receptor tyrosine kinase alleviates acute liver injury via inhibition of NLRP3 inflammasome activation in mice. Autophagy 2016, 12, 2326–2343.
- Bellan, M.; Sainaghi, P.P.; Minh, M.T.; Minisini, R.; Molinari, L.; Baldrighi, M.; Salmi, L.; Barbaglia, M.N.; Castello, L.M.; Ravanini, P.; et al. Gas6 as a predictor of esophageal varices in patients affected by hepatitis C virus related-chronic liver disease. Biomark. Med. 2018, 12, 27–34.
- Staufer, K.; Dengler, M.; Huber, H.; Marculescu, R.; Stauber, R.; Lackner, C.; Dienes, H.-P.; Kivaranovic, D.; Schachner, C.; Zeitlinger, M.; et al. The non-invasive serum biomarker soluble Axl accurately detects advanced liver fibrosis and cirrhosis. Cell Death Dis. 2017, 8, e3135.
- Zhu, C.; Wei, Y.; Wei, X. AXL receptor tyrosine kinase as a promising anti-cancer approach: Functions, molecular mechanisms and clinical applications. Mol. Cancer 2019, 18, 1–22.
- Holstein, E.; Binder, M.; Mikulits, W. Dynamics of Axl Receptor Shedding in Hepatocellular Carcinoma and Its Implication for Theranostics. Int. J. Mol. Sci. 2018, 19, 4111.
- Reichl, P.; Dengler, M.; Van Zijl, F.; Huber, H.; Führlinger, G.; Reichel, C.; Sieghart, W.; Peck-Radosavljevic, M.; Grubinger, M.; Mikulits, W. Axl activates autocrine transforming growth factor-β signaling in hepatocellular carcinoma. Hepatology 2015, 61, 930–941.
- Myers, K.V.; Amend, S.R.; Pienta, K.J. Targeting Tyro3, Axl and MerTK (TAM receptors): Implications for macrophages in the tumor microenvironment. Mol. Cancer 2019, 18, 1–14.
- Haider, C.; Hnat, J.; Wagner, R.; Huber, H.; Timelthaler, G.; Grubinger, M.; Coulouarn, C.; Schreiner, W.; Schlangen, K.; Sieghart, W.; et al. Transforming Growth Factor-β and Axl Induce CXCL5 and Neutrophil Recruitment in Hepatocellular Carcinoma. Hepatology 2018, 69, 222–236.
- Chai, Z.-T.; Zhang, X.-P.; Ao, J.-Y.; Zhu, X.-D.; Wu, M.-C.; Lau, W.Y.; Sun, H.-C.; Cheng, S.-Q. AXL Overexpression in Tumor-Derived Endothelial Cells Promotes Vessel Metastasis in Patients with Hepatocellular Carcinoma. Front. Oncol. 2021, 11.
- Shen, L.; Lei, S.; Zhang, B.; Li, S.; Huang, L.; Czachor, A.; Breitzig, M.; Gao, Y.; Huang, M.; Mo, X.; et al. Skipping of exon 10 in Axl pre-mRNA regulated by PTBP1 mediates invasion and metastasis process of liver cancer cells. Theranostics 2020, 10, 5719–5735.
- Li, X.-Y.; Wen, J.-Y.; Jia, C.-C.; Wang, T.-T.; Li, X.; Dong, M.; Lin, Q.; Chen, Z.-H.; Ma, X.-K.; Wei, L.; et al. MicroRNA-34a-5p enhances sensitivity to chemotherapy by targeting AXL in hepatocellular carcinoma MHCC-97L cells. Oncol. Lett. 2015, 10, 2691–2698.
- Sauzay, C.; Petit, A.; Bourgeois, A.-M.; Barbare, J.-C.; Chauffert, B.; Galmiche, A.; Houessinon, A. Alpha-foetoprotein (AFP): A multi-purpose marker in hepatocellular carcinoma. Clin. Chim. Acta 2016, 463, 39–44.
- Song, X.; Wu, A.; Ding, Z.; Liang, S.; Zhang, C. Soluble Axl Is a Novel Diagnostic Biomarker of Hepatocellular Carcinoma in Chinese Patients with Chronic Hepatitis B Virus Infection. Cancer Res. Treat. 2020, 52, 789–797.
- Dengler, M.; Staufer, K.; Huber, H.; Stauber, R.; Bantel, H.; Weiss, K.H.; Starlinger, P.; Pock, H.; Plachky, P.K.; Gotthardt, D.N.; et al. Soluble Axl is an accurate biomarker of cirrhosis and hepatocellular carcinoma development: Results from a large scale multicenter analysis. Oncotarget 2017, 8, 46234–46248.
- Trojan, J. Cabozantinib for the Treatment of Advanced Hepatocellular Carcinoma: Current Data and Future Perspectives. Drugs 2020, 80, 1203–1210.
- Hayashi, M.; Abe, K.; Fujita, M.; Takahashi, A.; Hashimoto, Y.; Ohira, H. Serum Gas6 and Axl as non-invasive biomarkers of advanced histological stage in primary biliary cholangitis. Hepatol. Res. 2020, 50, 1337–1346.
- Khamko, R.; Daduang, J.; Settasatian, C.; Limpaiboon, T. OPCML Exerts Antitumor Effects in Cholangiocarcinoma via AXL/STAT3 Inactivation and Rho GTPase Down-regulation. Cancer Genom.-Proteom. 2021, 18, 771–780.
- Jung, D.E.; Park, S.B.; Kim, K.; Kim, C.; Song, S.Y. CG200745, an HDAC inhibitor, induces anti-tumour effects in cholangiocarcinoma cell lines via miRNAs targeting the Hippo pathway. Sci. Rep. 2017, 7, 10921.
- Fujimori, T.; Kato, K.; Fujihara, S.; Iwama, H.; Yamashita, T.; Kobayashi, K.; Kamada, H.; Morishita, A.; Kobara, H.; Mori, H.; et al. Antitumor effect of metformin on cholangiocarcinoma: In vitro and in vivo studies. Oncol. Rep. 2015, 34, 2987–2996.