Gut-associated lymphoid tissue is one of the most diverse and complex immune compartments in the human body. The subepithelial compartment of the gut consists of immune cells of innate and adaptive immunity, non-hematopoietic mesenchymal cells, and stem cells of different origins, and is organized into secondary (and even tertiary) lymphoid organs, such as Peyer’s patches, cryptopatches, and isolated lymphoid follicles. The function of isolated lymphoid follicles is multifaceted; they play a role in the development and regeneration of the large intestine and the maintenance of (immune) homeostasis. Isolated lymphoid follicles are also extensively associated with the epithelium and its conventional and non-conventional immune cells; hence, they can also function as a starting point or maintainer of pathological processes such as inflammatory bowel diseases or colorectal carcinogenesis. These relationships can significantly affect both physiological and pathological processes of the intestines.
- colitis
- colorectal cancer
- isolated lymphoid follicles
- innate lymphoid cells
1. Isolated Lymphoid Follicles in Colonic Inflammation
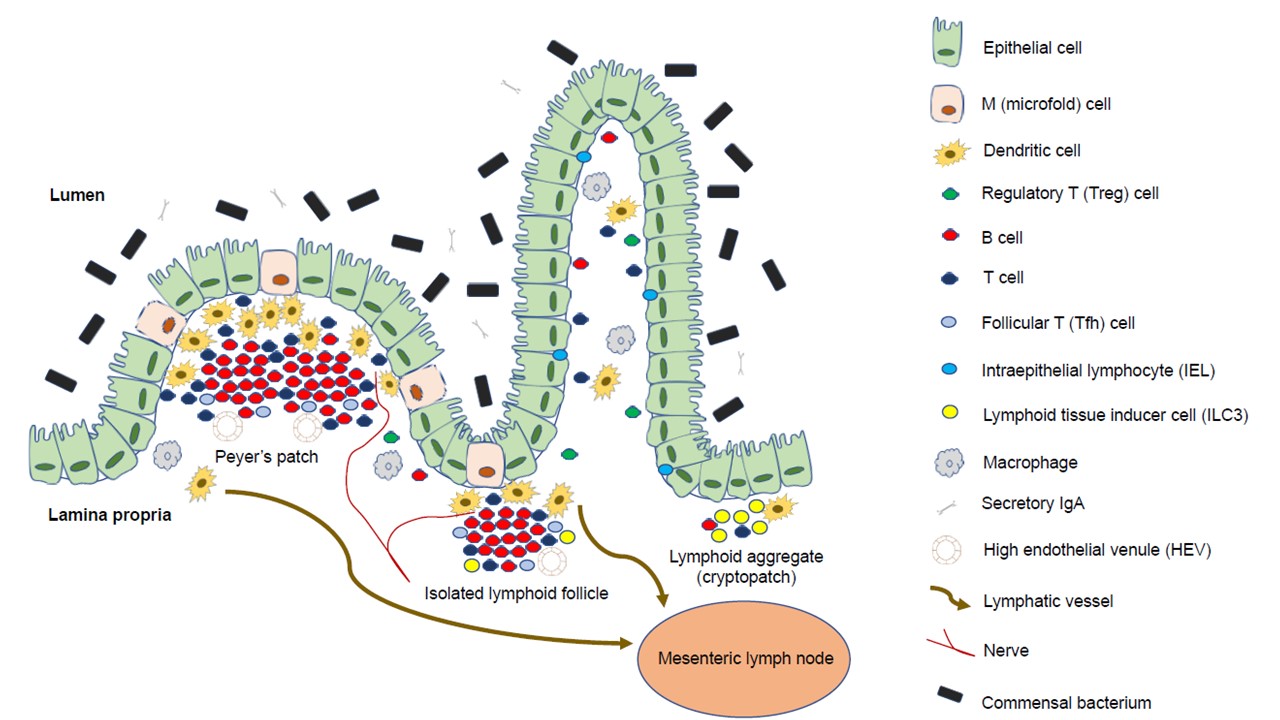
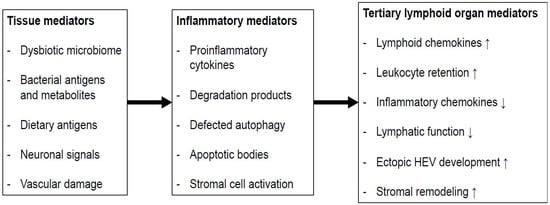
2. Isolated Lymphoid Follicles and Tertiary Lymphoid Organs in Colorectal Carcinogenesis
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10020226
References
- Luo, W.; Tian, L.; Tan, B.; Shen, Z.; Xiao, M.; Wu, S.; Meng, X.; Wu, X.; Wang, X. Update: Innate lymphoid cells in inflammatory bowel disease. Dig. Dis Sci. 2022, 67, 56–66.
- Giuffrida, P.; Corazza, G.R.; Di Sabatino, A. Old and new lymphocyte players in inflammatory bowel disease. Dig. Dis Sci. 2018, 63, 277–288.
- Drayton, D.L.; Liao, S.; Mounzer, R.H.; Ruddle, N.H. Lymphoid organ development: From ontogeny to neogenesis. Nat. Immunol. 2006, 7, 344–353.
- Sepahi, A.; Liu, Q.; Friesen, L.; Kim, C.H. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. 2021, 14, 317–330.
- Zhou, W.; Sonnenberg, G.F. Activation and suppression of group 3 innate lymphoid cells in the gut. Trends Immunol. 2020, 41, 721–733.
- Wu, Y.; Shen, J. Innate lymphoid cells in Crohn’s disease. Front. Immunol. 2020, 11, 554880.
- Diefenbach, A.; Gnafakis, S.; Shomrat, O. Innate lymphoid cell-epithelial cell modules sustain intestinal homeostasis. Immunity 2020, 52, 452–463.
- Saez, A.; Gomez-Bris, R.; Herrero-Fernandez, B.; Mingorance, C.; Rius, C.; Gonzalez-Granado, J.M. Innate lymphoid cells in intestinal homeostasis and inflammatory bowel disease. Int. J. Mol. Sci. 2021, 22, 7618.
- Zitti, B.; Bryceson, Y.T. Natural killer cells in inflammation and autoimmunity. Cytokine Growth Factor Rev. 2018, 42, 37–46.
- Hosomi, S.; Grootjans, J.; Tschurtschenthaler, M.; Krupka, N.; Matute, J.D.; Flak, M.B.; Martinez-Naves, E.; Gomez Del Moral, M.; Glickman, J.N.; Ohira, M.; et al. Intestinal epithelial cell endoplasmic reticulum stress promotes MULT1 up-regulation and NKG2D-mediated inflammation. J. Exp. Med. 2017, 214, 2985–2997.
- Gwela, A.; Siddhanathi, P.; Chapman, R.W.; Travis, S.; Powrie, F.; Arancibia-Cárcamo, C.V.; Geremia, A. Th1 and innate lymphoid cells accumulate in primary sclerosing cholangitis-associated inflammatory Bowel disease. J. Crohns Colitis. 2017, 11, 1124–1134.
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755.
- Tsai, P.Y.; Zhang, B.; He, W.Q.; Zha, J.M.; Odenwald, M.A.; Singh, G.; Tamura, A.; Shen, L.; Sailer, A.; Yeruva, S.; et al. IL-22 upregulates epithelial claudin-2 to drive diarrhea and enteric pathogen clearance. Cell Host Microbe 2017, 21, 671–681.e4.
- Li, J.; Shi, W.; Sun, H.; Ji, Y.; Chen, Y.; Guo, X.; Sheng, H.; Shu, J.; Zhou, L.; Cai, T.; et al. Activation of DR3 signaling causes loss of ILC3s and exacerbates intestinal inflammation. Nat. Commun. 2019, 10, 3371.
- Forkel, M.; van Tol, S.; Höög, C.; Michaëlsson, J.; Almer, S.; Mjösberg, J. Distinct alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established Crohn’s disease and ulcerative colitis. J. Crohns Colitis. 2019, 13, 67–78.
- Mazzurana, L.; Bonfiglio, F.; Forkel, M.; D’Amato, M.; Halfvarson, J.; Mjösberg, J. Crohn’s disease is associated with activation of circulating innate lymphoid cells. Inflamm Bowel Dis. 2021, 27, 1128–1138.
- Neurath, M.F. Targeting immune cell circuits and trafficking in inflammatory bowel disease. Nat. Immunol. 2019, 20, 970–979.
- Krauss, E.; Agaimy, A.; Neumann, H.; Schulz, U.; Kessler, H.; Hartmann, A.; Neurath, M.F.; Raithel, M.; Mudter, J. Characterization of lymphoid follicles with red ring signs as first manifestation of early Crohn’s disease by conventional histopathology and confocal laser endomicroscopy. Int. J. Clin. Exp. Pathol. 2012, 5, 411–421.
- Gullberg, E.; Söderholm, J.D. Peyer’s patches and M cells as potential sites of the inflammatory onset in Crohn’s disease. Ann. N Y Acad. Sci. 2006, 1072, 218–232.
- Van Kruiningen, H.J.; Colombel, J.F. The forgotten role of lymphangitis in Crohn’s disease. Gut 2008, 57, 1–4.
- Sura, R.; Colombel, J.F.; Van Kruiningen, H.J. Lymphatics, tertiary lymphoid organs and the granulomas of Crohn’s disease: An immunohistochemical study. Aliment. Pharm. Ther. 2011, 33, 930–939.
- McNamee, E.N.; Rivera-Nieves, J. Ectopic tertiary lymphoid tissue in inflammatory Bowel disease: Protective or provocateur? Front. Immunol. 2016, 7, 308.
- Neurath, M.F. Current and emerging therapeutic targets for IBD. Nat. Rev. Gastroenterol Hepatol. 2017, 14, 269–278.
- Smillie, C.S.; Biton, M.; Ordovas-Montanes, J.; Sullivan, K.M.; Burgin, G.; Graham, D.B.; Herbst, R.H.; Rogel, N.; Slyper, M.; Waldman, J.; et al. Intra- and inter-cellular rewiring of the human colon during ulcerative colitis. Cell 2019, 178, 714–730.e22.
- Smids, C.; Horjus Talabur Horje, C.S.; Drylewicz, J.; Roosenboom, B.; Groenen, M.J.M.; van Koolwijk, E.; van Lochem, E.G.; Wahab, P.J. Intestinal T Cell Profiling in Inflammatory Bowel Disease: Linking T Cell Subsets to Disease Activity and Disease Course. J. Crohns Colitis. 2018, 12, 465–475.
- Horjus Talabur Horje, C.S.; Smids, C.; Meijer, J.W.; Groenen, M.J.; Rijnders, M.K.; van Lochem, E.G.; Wahab, P.J. High endothelial venules associated with T cell subsets in the inflamed gut of newly diagnosed inflammatory bowel disease patients. Clin. Exp. Immunol. 2017, 188, 163–173.
- Kinchen, J.; Chen, H.H.; Parikh, K.; Antanaviciute, A.; Jagielowicz, M.; Fawkner-Corbett, D.; Ashley, N.; Cubitt, L.; Mellado-Gomez, E.; Attar, M.; et al. Structural remodeling of the human colonic mesenchyme in inflammatory Bowel disease. Cell 2018, 175, 372–386.e17.
- Mörbe, U.M.; Jørgensen, P.B.; Fenton, T.M.; von Burg, N.; Riis, L.B.; Spencer, J.; Agace, W.W. Human gut-associated lymphoid tissues (GALT); diversity, structure, and function. Mucosal Immunol. 2021, 14, 793–802.
- Chiba, M.; Yamano, H.; Fujiwara, K.; Abe, T.; Iizuka, M.; Watanabe, S. Lymph folliculitis in ulcerative colitis. Scand, J. Gastroenterol. 2001, 36, 332–336.
- Lochner, M.; Ohnmacht, C.; Presley, L.; Bruhns, P.; Si-Tahar, M.; Sawa, S.; Eberl, G. Microbiota-induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORgamma t and LTi cells. J. Exp. Med. 2011, 208, 125–134.
- Isene, R.; Bernklev, T.; Høie, O.; Munkholm, P.; Tsianos, E.; Stockbrügger, R.; Odes, S.; Palm, Ø.; Småstuen, M.; Moum, B. EC-IBD study group. extraintestinal manifestations in Crohn’s disease and ulcerative colitis: Results from a prospective, population-based European inception cohort. Scand J. Gastroenterol. 2015, 50, 300–305.
- Belvedere, A.; Scoglio, R.; Viola, A.; Costantino, G.; Sitibondo, A.; Muscianisi, M.; Inferrera, S.; Alibrandi, A.; Fries, W. A real world investigation on prevalence, clinical features, and therapy of inflammatory bowel disease in the city of Messina, Italy. Acta Biomed. 2021, 92, e2021161.
- Schaubeck, M.; Clavel, T.; Calasan, J.; Lagkouvardos, I.; Haange, S.B.; Jehmlich, N.; Basic, M.; Dupont, A.; Hornef, M.; von Bergen, M.; et al. Dysbiotic gut microbiota causes transmissible Crohn’s disease-like ileitis independent of failure in antimicrobial defence. Gut 2016, 65, 225–237.
- McNamee, E.N.; Masterson, J.C.; Jedlicka, P.; Collins, C.B.; Williams, I.R.; Rivera-Nieves, J. Ectopic lymphoid tissue alters the chemokine gradient, increases lymphocyte retention and exacerbates murine ileitis. Gut 2013, 62, 53–62.
- Zhao, L.; Zhang, X.; Zuo, T.; Yu, J. The composition of colonic commensal bacteria according to anatomical localization in colorectal cancer. Engineering 2017, 3, 90–97.
- Maoz, A.; Dennis, M.; Greenson, J.K. The Crohn’s-like lymphoid reaction to colorectal cancer-tertiary lymphoid structures with immunologic and potentially therapeutic relevance in colorectal cancer. Front. Immunol. 2019, 10, 1884.
- Väyrynen, J.P.; Sajanti, S.A.; Klintrup, K.; Mäkelä, J.; Herzig, K.H.; Karttunen, T.J.; Tuomisto, A.; Mäkinen, M.J. Characteristics and significance of colorectal cancer associated lymphoid reaction. Int. J. Cancer. 2014, 134, 2126–2135.
- Di Caro, G.; Bergomas, F.; Grizzi, F.; Doni, A.; Bianchi, P.; Malesci, A.; Laghi, L.; Allavena, P.; Mantovani, A.; Marchesi, F. Occurrence of tertiary lymphoid tissue is associated with T-cell infiltration and predicts better prognosis in early-stage colorectal cancers. Clin. Cancer Res. 2014, 20, 2147–2158.
- Rozek, L.S.; Schmit, S.L.; Greenson, J.K.; Tomsho, L.P.; Rennert, H.S.; Rennert, G.; Gruber, S.B. Tumor-infiltrating lymphocytes, Crohn’s-like lymphoid reaction, and survival from colorectal cancer. J. Natl. Cancer Inst. 2016, 108, djw027.
- Ogino, S.; Nosho, K.; Irahara, N.; Meyerhardt, J.A.; Baba, Y.; Shima, K.; Glickman, J.N.; Ferrone, C.R.; Mino-Kenudson, M.; Tanaka, N.; et al. Lymphocytic reaction to colorectal cancer is associated with longer survival, independent of lymph node count, microsatellite instability, and CpG island methylator phenotype. Clin. Cancer Res. 2009, 15, 6412–6420.
- Ueno, H.; Hashiguchi, Y.; Shimazaki, H.; Shinto, E.; Kajiwara, Y.; Nakanishi, K.; Kato, K.; Maekawa, K.; Miyai, K.; Nakamura, T.; et al. Objective criteria for crohn-like lymphoid reaction in colorectal cancer. Am. J. Clin. Pathol. 2013, 139, 434–441.
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Pommerencke, T.; von Knebel Doeberitz, M.; Schirmacher, P.; Weitz, J.; Grabe, N.; Jäger, D. The localization and density of immune cells in primary tumors of human metastatic colorectal cancer shows an association with response to chemotherapy. Cancer Immun. 2009, 9, 1.
- Halama, N.; Michel, S.; Kloor, M.; Zoernig, I.; Benner, A.; Spille, A.; Pommerencke, T.; von Knebel, D.M.; Folprecht, G.; Luber, B.; et al. Localization and density of immune cells in the invasive margin of human colorectal cancer liver metastases are prognostic for response to chemotherapy. Cancer Res. 2011, 71, 5670–5677.
- Wouters, M.C.A.; Nelson, B.H. Prognostic significance of tumor-infiltrating B cells and plasma cells in human cancer. Clin. Cancer Res. 2018, 24, 6125–6135.
- Tsou, P.; Katayama, H.; Ostrin, E.J.; Hanash, S.M. The emerging role of B cells in tumor immunity. Cancer Res. 2016, 76, 5597–5601.
- Yuen, G.J.; Demissie, E.; Pillai, S. B lymphocytes and cancer: A love-hate relationship. Trends Cancer. 2016, 2, 747–757.
- Linnebacher, M.; Maletzki, C. Tumor-infiltrating B cells: The ignored players in tumor immunology. Oncoimmunology 2012, 1, 1186–1188.
- Van Nierop, K.; de Groot, C. Human follicular dendritic cells: Function, origin and development. Semin. Immunol. 2002, 14, 251–257.
- Mungenast, F.; Meshcheryakova, A.; Beer, A.; Salzmann, M.; Tamandl, D.; Gruenberger, T.; Pietschmann, P.; Koperek, O.; Birner, P.; Kirsch, I.; et al. The immune phenotype of isolated lymphoid structures in non-tumorous colon mucosa encrypts the information on pathobiology of metastatic colorectal cancer. Cancers 2020, 12, 3117.
- Gatto, D.; Brink, R. The germinal center reaction. J. Allergy Clin. Immunol. 2010, 126, 898–907.
- Stebegg, M.; Kumar, S.D.; Silva-Cayetano, A.; Fonseca, V.R.; Linterman, M.A.; Graca, L. Regulation of the germinal center response. Front. Immunol. 2018, 9, 2469.
- Miyasaka, M.; Tanaka, T. Lymphocyte trafficking across high endothelial venules: Dogmas and enigmas. Nat. Rev. Immunol. 2004, 4, 360–370.
- Mechtcheriakova, D.; Svoboda, M.; Meshcheryakova, A.; Jensen-Jarolim, E. Activation-induced cytidine deaminase (AID) linking immunity, chronic inflammation, and cancer. Cancer Immunol. Immunother. 2012, 61, 1591–1598.
- Bannard, O.; Cyster, J.G. Germinal centers: Programmed for affinity maturation and antibody diversification. Curr. Opin. Immunol. 2017, 45, 21–30.
- Nelson, B.H. CD20+ B cells: The other tumor-infiltrating lymphocytes. J. Immunol. 2010, 185, 4977–4982.
- Boothby, M.R.; Hodges, E.; Thomas, J.W. Molecular regulation of peripheral B cells and their progeny in immunity. Genes Dev. 2019, 33, 26–48.
- Seifert, M.; Küppers, R. Human memory B cells. Leukemia 2016, 30, 2283–2292.
- Shaheen, S.; Guddati, A.K. Secondary mucosa-associated lymphoid tissue (MALT) lymphoma of the colon. Med. Oncol. 2013, 30, 502.
- Buettner, M.; Lochner, M. Development and function of secondary and tertiary lymphoid organs in the small intestine and the colon. Front. Immunol. 2016, 7, 342.
- Knoop, K.A.; Newberry, R.D. Isolated lymphoid follicles are dynamic reservoirs for the induction of intestinal IgA. Front. Immunol. 2012, 3, 84.
- Pearson, C.; Uhlig, H.H.; Powrie, F. Lymphoid microenvironments and innate lymphoid cells in the gut. Trends Immunol. 2012, 33, 289–296.
- Pabst, O.; Herbrand, H.; Worbs, T.; Friedrichsen, M.; Yan, S.; Hoffmann, M.W.; Körner, H.; Bernhardt, G.; Pabst, R.; Förster, R. Cryptopatches and isolated lymphoid follicles: Dynamic lymphoid tissues dispensable for the generation of intraepithelial lymphocytes. Eur. J. Immunol. 2005, 35, 98–107.
- Pagès, F.; Mlecnik, B.; Marliot, F.; Bindea, G.; Ou, F.S.; Bifulco, C.; Lugli, A.; Zlobec, I.; Rau, T.T.; Berger, M.D.; et al. International validation of the consensus immunoscore for the classification of colon cancer: A prognostic and accuracy study. Lancet 2018, 391, 2128–2139.
- Buchan, S.L.; Rogel, A.; Al-Shamkhani, A. The immunobiology of CD27 and OX40 and their potential as targets for cancer immunotherapy. Blood 2018, 131, 39–48.
- Buchan, S.L.; Fallatah, M.; Thirdborough, S.M.; Taraban, V.Y.; Rogel, A.; Thomas, L.J.; Penfold, C.A.; He, L.Z.; Curran, M.A.; Keler, T.; et al. PD-1 blockade and CD27 stimulation activate distinct transcriptional programs that synergize for CD8+ T-Cell-driven antitumor immunity. Clin. Cancer Res. 2018, 24, 2383–2394.
- Hernandez, P.; Gronke, K.; Diefenbach, A. A catch-22: Interleukin-22 and cancer. Eur. J. Immunol. 2018, 48, 15–31.
- Penny, H.A.; Hodge, S.H.; Hepworth, M.R. Orchestration of intestinal homeostasis and tolerance by group 3 innate lymphoid cells. Semin. Immunopathol. 2018, 40, 357–370.
- Bergmann, H.; Roth, S.; Pechloff, K.; Kiss, E.A.; Kuhn, S.; Heikenwälder, M.; Diefenbach, A.; Greten, F.R.; Ruland, J. Card9-dependent IL-1β regulates IL-22 production from group 3 innate lymphoid cells and promotes colitis-associated cancer. Eur. J. Immunol. 2017, 47, 1342–1353.