Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Transforming growth factor (TGF)-β is the name for a superfamily of proteins, including myostatin, that functions in the body to affect growth and to stimulate the inflammatory response along with other functions covered elsewhere. TGF-β in skeletal muscle not only contributed to muscle fibrosis in Duchenne Muscular Dystrophy (DMD) disease, but also promoted skeletal muscle atrophy by decreasing muscle fiber diameter and amounts of heavy chain myosin (MHC) in muscle tissue.
- transforming growth factor-beta
- skeletal muscle wasting
- reactive oxygen species
- metastatic cancer
- pediatric burns
1. The Role of Bone
In 2015, Waning et al. [10] described changes to the bone microenvironment in both mouse models and in patients with bone metastases from breast and lung cancer. The work of this group identified that bony metastases from cancers resulted in inflammation, with resultant liberation of TGF-β from bone. The result of this release was the generation of ROS in the sarcoplasmic reticulum of skeletal muscle, where it interacts with the ryanodine receptor, oxidizing it and causing a calcium leak resulting in muscle weakness. The effects of TGF-β on the ryanodine receptor could be reversed by use of either a TGF-β1 receptor blocker or the anti-resorptive agent zoledronate. This study confirmed the work of Abrigo et al. [7] in identifying ROS generation as a mode of action leading to muscle weakness in metastatic cancer. Another piece of the puzzle, the mechanics of the release of TGF-β from resorbing bone, was supplied by the work of Dallas et al. [11]. This group reported that TGF-β was stored in the bone extracellular matrix bound to Latent TGF-β Binding Protein-1 and that osteoclasts, with the aid of matrix metalloproteinases (MMP)-2 and -9, can cleave this binding protein from TGF-β, liberating it from the extracellular matrix during resorption. Figure 1 illustrates the known pathway of TGF-β release from bone. What was, at the time, unclear was whether this TGF-β liberation from bone with paracrine catabolic effects on muscle was unique to metastatic cancers. As it turns out, Pin et al. [12] subsequently described muscle wasting in cancer without metastases, inasmuch as IL-6, tumor necrosis factor-alpha (TNF-α), interferon-gamma (IFN-¥) and IL-1-β are chronically elevated in blood, peripheral tissues, and the central nervous system of patients with cancer cachexia. These cytokines can be produced by either the immune cells or tumor cells. Moreover, myostatin, a member of the TGF-β family of proteins, and one that inhibits muscle protein synthesis, has a catabolic effect on bone by inhibiting osteoblast differentiation by decreasing the osteocyte-derived production of exosomal microRNA (mIR) 218 [13]. Myostatin also increases RANK Ligand activity in stimulating osteoclast formation. To compound matters, Chen et al. [14] showed that the inhibition of activin receptors and of myostatin inhibited the Smad 2/3 pathway, producing muscle hypertrophy. The driving factor in the muscle hypertrophy was an increase in muscle protein synthesis and a reduction in ubiquitin ligase activity—findings seen with the inhibition of TGF-β release from bone. Thus, the relationship between the TGF-β molecule liberated from bone during resorption, and muscle myostatin activity, remains unclear.
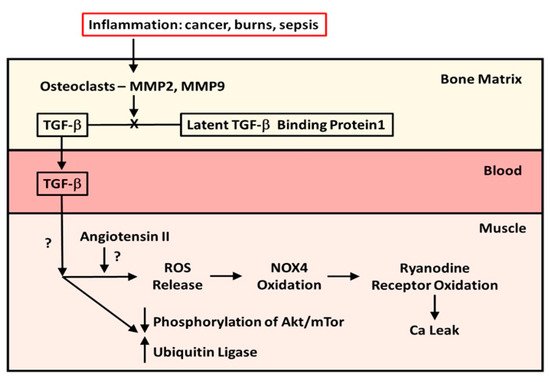
Figure 1. The mechanism and effect of release of TGF-β from bone matrix. Diagram of the cleavage of TGF-β from Latent TGF-β Binding Protein-1 by osteoclasts in the bone matrix during resorption and the effects of TGF-β released from bone during resorption on muscle protein metabolism in patients with cancer metastatic to bone and in pediatric burn patients.
However, it is known that both TGF-β1 and myostatin bind to the same receptors, which, when bound, will phosphorylate the Smad 2/3 signaling pathway, resulting in stimulation of the ubiquitin ligase muscle protein catabolic pathway and suppression of the Akt/mTor muscle protein anabolic pathway.
Not only is muscle wasting produced by cancer metastases and cancer without metastases, but chemotherapeutic agents, such as carboplatin [15] and cisplatin [16], are capable of producing bone loss and consequent muscle wasting with zoledronate preventing the muscle wasting in both instances. The authors of these papers did not specify that activation of TGF-β was involved in the pathogenesis of the muscle wasting.
Expansion of the Role of Bone-Liberated TGF-β to Other Musculoskeletal Conditions
At about the same time as Waning et al. [10] were carrying out their studies, our group was reviewing data from a previous randomized double-blind placebo-controlled trial (RCT) of a bisphosphonate, pamidronate, in blocking bone resorption following the well-known burn-induced systemic inflammatory response [17]. The data obtained had clearly indicated that anti-resorptive agents were safe and effective in preventing bone loss following severe burns in children [17]. When we identified one-third of our RCT patients in a database of pediatric burn patients who completed stable isotope studies of muscle protein kinetics during the first 30 days post-burn, we found that those patients who had received the bisphosphonate did not experience muscle wasting compared to those subjects who had received a placebo, and in contrast to the expected negative muscle protein balance normally seen with burns, these children were in positive muscle protein balance [18]. With both Waning et al. [10] and our group presenting our results at the same meeting in China in 2014, we speculated that the burn patients were being affected by a mechanism similar to what was seen in the mice and patients with metastatic cancer. It was not until 2019 that Pin et al. [19] published the results of in vitro studies utilizing murine C2C12 myoblast cultures containing serum from burned subjects who received either bisphosphonate or placebo or from normal unburned pediatric subjects. They reported that myoblast cultures containing serum from burn patients receiving placebo showed diminished myotube size that was rescued by serum in culture from bisphosphonate-treated patients. Further, placebo-treated burn patient serum in myoblast cultures stimulated the ubiquitin ligase catabolic pathway and suppressed phosphorylation of the anabolic Akt/mTor pathway, while the reverse occurred in cultures with bisphosphonate-containing serum from burn patients. When myoblast cultures with placebo-treated serum were cultured with anti-TGF-β 1–3 antibody, the rescue of myotube size was comparable in magnitude to that in cultures with bisphosphonate-containing serum, indicating that TGF-β was responsible for the reduction in myotube size in pediatric burns, and that preventing bone resorption was sufficient to preserve myotube size in culture and to stimulate the anabolic Akt/mTor pathway while suppressing the catabolic ubiquitin ligase pathway. These latter findings are consistent with part of the mechanism of action of TGF-β on muscle reported by Abrigo et al. [7]. Figure 1 also summarizes what is known of the effects of TGF-β on skeletal muscle.
The findings in pediatric burn patients, while not addressing the generation of reactive oxygen species seen with cancer metastatic to bone, did demonstrate the influences of TGF-β on the muscle catabolic pathways and suppression of phosphorylation of the anabolic pathway.
Thus, two very distinct groups of patients, mature adults with cancer with or without metastases, and previously healthy children and adolescents with acute, severe burn injury, have exhibited similar, if not identical, mechanisms of muscle wasting that were influenced by the release of TGF-β from bone. This observation suggests that the release of TGF-β from bone during resorption helps bone control muscle mass and strength. There may also be therapeutic implications of this finding so that factors increasing bone resorption, such as inflammation and possibly immobilization, could be counteracted by the use of anti-resorptive agents, reducing the bone release of TGF-β with consequent sparing of muscle mass and strength.
A potential third disorder in which TGF-β release may be involved was reported by Jude et al. [20], showing that inhibition of TGF-β1 in septic rats using the TGF-β inhibitor LY364947 protected the diaphragm muscle from sepsis-induced weakness and wasting. Additionally, in vitro studies on old and young muscle from mice by Carlson et al. [21] demonstrated that, at least in mice, old muscle produces excess TGF-β1, but not myostatin, and the former induces high levels of TGF-β phosphorylated Smad3 in muscle satellite cells, interfering with the regenerative capacity of these muscle stem cells. This finding suggests that skeletal muscle from old mice may develop sarcopenia as a result of the increased expression of TGF-β. However, it raises the question as to whether bone resorption releases TGF-β as well, and, if so, what is the relationship between bone TGF-β and muscle TGF-β in elderly humans? Does the presence of TGF-β from bone induce muscle to express more TGF-β? A more recent paper by Zhang et al. [22] that identified an intrinsic TGF-β inhibitor in liver, hemojuvelin, which could suppress TGF-β in muscle from both Duchenne muscular dystrophy and from aging mice, is also consistent with increased muscle TGF-β production in aging animals.
2. Evidence Suggesting That Immobilization Plays a Role in TGF-β-Mediated Muscle Wasting
We do not have evidence implicating bone-released TGF-β’s involvement in immobilization to date. However, a recent study by Gugala et al. [31] provides suggestive information. Studies of a rat model of critical illness myopathy developed by Llano Diez et al. [32] have shown that by pharmacologically immobilizing rats and mechanically ventilating them for 10 days, a model that reproduces the effects of immobilization and the mechanical ventilation of patients in intensive care for COVID-19 respiratory disease, the loss of appendicular skeletal muscle mass and, in particular, myosin, occurs in tight correlation with the loss of trabecular bone, as determined by serial micro computed tomography of the femurs of rats studied from 0–10 days post-immobilization. While immobilization is the main feature of this model, it is difficult to discount a role of inflammation that may occur as a result of mechanical ventilation. This is because mechanical ventilation itself causes an inflammatory response by upregulating the NLRP3 inflammasome [33], resulting in the increased production of IL-1 by the monocytes and macrophages of the innate immune system. Therefore, the model is not entirely “clean”, in a manner similar to burn injury. However, the tightness of the correlation between muscle and bone loss in this experimental setting is suggestive that bone resorption is involved in the pathogenesis of muscle wasting in this model. The definitive experiment would involve the use of bisphosphonates or other anti-resorptive agents to block the bone loss and then evaluate the muscle wasting. However, studies of atrophic muscle fibers in this condition have been reported to express TGF-β ligands [34]. Similarly, studies of sarcopenia have reported an increase in circulating TGF-β1, phosphorylated Smad 3, and myostatin, again suggesting the involvement of TGF-β1 in these conditions [35]. Yet, it is unclear whether muscle myostatin is upregulated by TGF-β released from bone, although, as we have seen [21], this may not occur in aging, sarcopenic muscle. However, this relationship has not been studied in other conditions, so it is not clear that this is categorically the case.
This entry is adapted from the peer-reviewed paper 10.3390/ijms23031167
This entry is offline, you can click here to edit this entry!