Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
The mammalian or mechanistic target of rapamycin (mTOR) pathway plays a crucial role in regulation of cell survival, metabolism, growth and protein synthesis in response to upstream signals in both normal physiological and pathological conditions, especially in cancer.
- mTOR
- cancer
- inhibitor
1. mTORC1 and mTORC2
mTOR is a serine/threonine kinase, which is attributed to the phosphoinositide 3-kinase related protein kinase (PIKK) super family, and was first discovered from a genetic screening for rapamycin-resistant mutations in yeast Saccharomyces cerevisiase [1][2]. In mammalian cells, mTOR mainly acts through its two evolutionarily conserved complexes, mTORC1 and mTORC2, which share some common subunits, such as the mTOR kinase, the mammalian lethal with SEC13 protein 8 (mLST8), dishevelled, EGL-10 and pleckstrin (DEP) domain-containing mTOR-interacting protein (DEPTOR), telomere maintenance 2 (Tel2) and Tel2-interacting protein 1(Tti1) complex as shown in Figure 1.
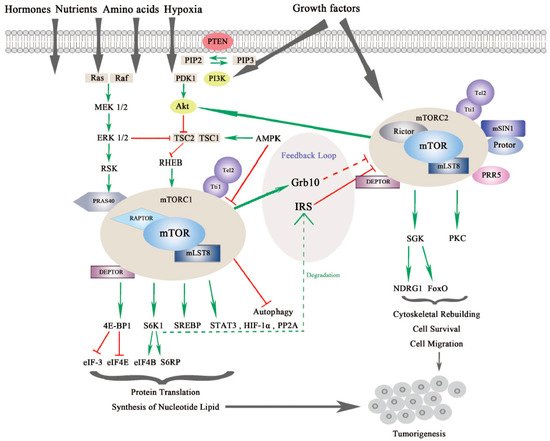
Figure 1. The mammalian or mechanistic target of rapamycin (mTOR) complexes and signaling pathway of mTORC1 and mTORC2. mTORC1 is responsive to nutrients, hormones, amino acids, hypoxia and growth factors, while mTORC2 responds to growth factors. mTORC1 and mTORC2 share common subunits of mTOR kinase, mLST8, DEPTOR (DEP domain-containing mTOR-interacting protein), Tel 2 and Tti 1. mTORC1 additionally binds with RAPTOR (Regulatory-associated protein of mTOR) and PRAS40 (Proline-rich substrate of 40 kDa), and mTORC2 combines with RICTOR and mSIN1 (Mammalian stress-activated protein kinase interacting protein 1) as well as Protor and PRR5 (Proline-rich protein 5). mTORC1 is regulated by PI3K/Akt (Phosphoinositide 3-kinase/serine-threonine protein kinase) and Ras-MAPK (Mitogen activated protein kinase) signaling pathways. mTORC1 regulates protein translation and synthesis of nucleotide lipid via 4E-BP1 and S6K1 and downstream effectors. mTORC1 also activates STAT3 (Signal transducer and activator of transcription), HIF-1α (Hypoxia-inducible factor 1α) and PP2A (Protein phosphatase 2A) in tumorigenesis. mTORC2 regulates SGK (Serum glucose kinase) and PKC (Protein kinase C) to promote cell survival, cytoskeleton reorganization and cell migration. mTORC2 is negatively modulated by mTORC1 via different feedback loops mediated by IRS (insulin receptor substrate) or Grb10. mTORC1 and mTORC2 can both contribute to turmorigenesis through different mechanisms [3][4].
mTORC1 and mTORC2 are different in the aspects of rapamycin sensitivity, specific binding components, subcellular localization, downstream substrates, and regulation [5]. mTORC1 is sensitive to rapamycin whereas mTORC2 is comparatively resistant to rapamycin [6]. In addition to the common binding subunits, mTORC1 and mTORC2 respectively harbor distinct components that contribute to the specificity of substrates, different subcellular localization, and specific regulation. mTORC1 also contains the regulatory-associated protein of mTOR (RAPTOR), which is a significant scaffolding protein in the mTORC1 assembly and its stability and regulation, and proline-rich substrate of 40 kDa (PRAS40) is a negative regulator of mTORC1 by releasing mTORC1 inhibition upon the activation of growth factors [7][8]. mTORC2 uniquely contains rapamycin-insensitive companion of mTOR (RICTOR) and the mammalian stress-activated protein kinase interacting protein 1 (mSIN1), both of which can mutually affect their protein levels and stabilize each other. Previous research has demonstrated that RICTOR is a scaffolding protein essential for the assembly, stability, substrate recognition, and subcellular localization activation of mTORC2. In addition, mSIN1, which is essential for plasma membrane localization of mTORC2, negatively regulates mTORC2 kinase activity [9][10]. Newly discovered interactors include Protein observed with RICTOR 1/2 (Protor-1/2), which are required for mTORC2 assembly and catalytic process, and Proline-Rich Protein (PRR) 5, which is necessary for mTOR activity and mTOR–RICTOR binding [11][12].
mTORC1 and mTORC2 have differing subcellular localization binding with their own respective, specific subunits, which also determine their distinct functions and independent regulations. mTORC1 is associated with endosomal and lysosomal membranes, where it interacts with its effectors. mTORC2 is affiliated with the plasma membrane, as well as ribosomal membranes, where it binds with its key substracts, AGC family kinases (subgroup of Ser/Thr protein kinases named after 3 representative families, the cAMP-dependent protein kinase (PKA), the cGMP-dependent protein kinase (PKG) and the protein kinase C (PKC) families), such as serum glucose kinase (SGK) isoforms and protein kinase C (PKC), which are essential for mTORC2 activation [13]. Both mTORC1 and mTORC2 play significant and differing roles in a variety of intracellular processes. They are regulated by various endogenous and exogenous stimuli, such as nutrients, growth factors, energy, hormones and hypoxia, and they can also affect glucose metabolism through different physiological mechanisms [14][15][16][17]. Generally, mTORC1 can phosphorylate its downstream effectors, such as eukaryotic translation initiation factor 4E binding protein 1 (4EBP1), S6 kinase (S6K), and sterol regulatory element-binding protein (SREBP), to motivate protein translation, synthesis of nucleotides and lipids, biogenesis of lysosomes, and to suppress the process of autophagy [18]. On the other hand, mTORC2 is more sensitive to extracellular growth factors though the molecular mechanism remains to be elucidated [19]. Upon activation, mTORC2 phosphorylates its downstream targets SGK and PKC, as mentioned previously, to intensify the signaling cascade [20]. mTORC2 mainly increases cytoskeletal rebuilding and cell migration, inhibits apoptosis and affects metabolism [21] (as shown in Figure 1).
2. Signaling of mTORC1
The mTOR signaling pathway is crucial in cell growth, proliferation and metabolism. mTORC1 is regulated by several signaling pathways including the PI3K/Akt pathway, the Ras-MAPK pathway, and some other intracellular factors (see Figure 1).
Activation of mTORC1 is primarily dependent on the PI3K/AKT pathway to respond to oncogenic growth factors or insulin [22]. Even though the second messenger phosphatidylinositol (3,4,5)-triphosphate (PIP3) binds and activates mTORC2 directly, mTORC1 can also be indirectly activated by PI3K through Akt. Akt is activated by phosphorylation at Ser473 by mTORC2 and at Thr308 by another serine-threonie kinase PDK1 (Phosphoinositide-dependent Kinase 1). Then, phosphorylation of tuberous sclerosis complex 2 (TSC2) by active Akt results in blockage of TSC2 and TSC1 combination [23][24][25]. The activator of mTORC1, Ras homolog enriched in brain (RHEB), which is negatively regulated by TSC1/2, is released by TSC to allow the activation of mTORC1 in lysosomes [26]. In addition, AKT can activate mTORC1 by phosphorylating and dissociating the inhibitor PRAS40 from RAPTOR independent of TSC1/2 [27].
Moreover, TSC2 can also be phosphorylated by extracellular signal-regulated kinases (ERKs) and ribosomal protein S6 kinase (RSK) from the Ras-MAPK signaling pathway, which results in inhibiting TSC1/2 and promoting RHEB-mediated mTORC1 activation. In addition, similar to AKT, PRAS40 can also be phosphorylated by RSK to release RAPTOR and activate mTORC1 [28][29][30].
mTORC1 is also responsive to fluctuations of cellular factors such as DNA damage, intracellular adenosine triphosphate (ATP), glucose, amino acids, and oxygen. Several signaling pathways that are responsive to DNA damage suppress mTORC1 via p53 target genes, leading to TSC2 activation: for example, 5′-AMP activated protein kinase β (AMPKβ) and phosphatase and tensin homolog on chromosome 10 (PTEN) [31]. Upon energy exhaustion, AMP kinase (AMPK), which is activated by low ATP/high AMP levels, promotes TSC1/2 complex formation and phosphorylates RAPTOR, leading to indirect inhibition of mTORC1 [32]. This outcome also implies that in a situation of energy shortage, AMP accumulation will cover the growth factor signals and suppress cellular replication. Through a sensing signal cascade of amino acids, mTORC1 can be positively regulated by amino acids, activation of which motivates the Rag complex to combine with RAPTOR. Along with this process, mTORC1 is recruited to the lysosomal surface [33][34]. Rag-GTPase, which is associated with RAPTOR and localizes mTORC1 to lysosomal membranes, is especially activated by arginine in lysosomes or by leucine in the cytoplasm [35][36][37][38].
Once activated, mTORC1 will transfer the signal to downstream effectors, such as 4EBP1 and S6K1, both of which are essential modulators of cap-dependent and cap-independent translation. After phosphorylation of 4EBP1 and S6K1 by mTORC1, the binding partners, eukaryotic initiation factor (eIF)-4E and eukaryotic initiation factor-3 (eIF-3), will be respectively liberated, facilitating initiating complex formation for translation and intensifying ribosome genesis [39]. In the following signal cascade, eIF-4E will form the eIF-4F complex and increase protein translation, which is significant for the G1-S phase transition. Upon low mTORC1 activity, 4E-BP1 is dephosphorylated, and protein translation is inhibited [40]. On the other side, eIF-4B and S6 ribosomal protein (S6RP) are phosphorylated by S6K1, which initiates protein translation and continues translation elongation [41][42]. Actually, mTORC1-related signals seem to prefer to affect the translation of oncogenic proteins involved in protein synthesis, invasion and metastasis [43]. Moreover, mTORC1 also regulates some other proteins such as hypoxia-inducible factor 1α (HIF-1α), protein phosphatase 2A (PP2A), glycogen synthase, and signal transducer and activator of transcription (STAT) 3, through which mTORC1 promotes biosynthesis of proteins, lipids and nucleotides in aberrant cells, tissue and organism growth in cancer [44][45][46][47][48][49].
3. Signaling of mTORC2
Although the regulatory mechanism of mTORC1 is well depicted, the regulators of mTORC2 are much less characterized. This is partly due to the difficulties in teasing apart the functional differences between mTORC1 and mTORC2 [6]. As we mentioned previously, through mSIN1, mTORC2 localizes at the plasma membrane where it binds with its substrates Akt, SGK and PKC. Notably, the localization of mTORC2 is significant for its regulation [9] (see Figure 1).
First, mSIN1 regulates mTORC2 depending on different mechanisms. mTORC2-Akt signaling can be sustained by a positive feedback loop from mSIN1 phosphorylation of Akt, whereas mSIN 1 phosphorylation by S6K1 at the same site suppresses mTORC2 activity [52][53][54]. On the other hand, recent research found that mSIN1 can also combine with Rb in the cytoplasm, which results in the inhibition of mTORC2 complex formation and Akt signaling [55].
Likewise, mTORC2 is regulated by PI3K/Akt, as well as by mTORC1 itself. PI3K activates mTORC2 to bind to ribosomes both in normal physiological and pathological conditions, such as cancer [56]. Akt, which is commonly found to be hyperactive in cancers, is an important substrate of mTORC2. Akt aggregates signals from PI3K/mTORC2 and PI3K/PDK1 to accelerate cell proliferation. Localization of Akt to the plasma membrane is regulated by PIP3, which is similar to mTORC2. Akt also activates mTORC1 signaling in addition to mTORC2, leading to a more complicated signal network [23]. In addition, mTORC2 is negatively modulated by mTORC1 via feedback loops. For example, the S6K1 promotes insulin receptor substrate (IRS) 1/2 degradation resulting in inhibition of mTORC2 and the PI3K/Akt pathway. Another feedback mechanism is through growth factor receptor-bound protein 10 (Grb10), which is positively modulated by mTORC1 [57][58][59].
For downstream effectors, serum and glucocorticoid kinase (SGK) and protein kinase C (PKC) are two key phosphorylation substrates of mTORC2. SGK substrates include N-myc downstream-regulated gene 1 protein (NDRG1) and Forkhead box family transcription factors (FoxO), which promote cell survival under oxygen or nutrient depletion conditions or in response to PI3K inhibition [60][61]. Through phosphorylation of different PKC family members, mTORC2 is reported to regulate cytoskeleton reorganization and cell movements involved in tumorigenesis [10][19][62][63] (See Figure 1).
2.4. mTOR Signaling in Cancer
Since mTOR signaling regulates fundamental activities including cell cycle, proliferation, growth, and survival, as well as protein synthesis and glucose metabolism, there is no doubt that mTOR has a close association with cancer. As reported, mTOR signaling is enhanced in various types of cancers. Data in solid tumors demonstrated that the mTOR signal is dysregulated in almost 30% of cancers and is one of the most frequently affected cascades in human cancers [64].
Activation of mTOR signaling in cancer mainly depends on three different levels of mechanisms: first, mutations in the mTOR gene lead to a constitutively hyperactive mTOR signaling cascade; second, mutations in the components of mTORC1 and mTORC2 result in activation of mTOR signaling; and lastly but most importantly, aberrant mTOR signaling can also result from mutations in upstream genes, that is, loss-of- function mutations in suppressor genes and gain-of-function mutations in oncogenes [3]. We discuss these mechanisms in the following text.
Mutation of mTOR, which is the core gene of the mTOR signaling and encodes the kinase, will directly lead to hyperactivation of mTOR signaling. A study utilizing public tumor genome sequencing data in 2014 reported that 33 mTOR mutations were found to contribute to the hyperactivation of mTOR signaling in various cancer types. Most of these mutations assemble in six different regions of the c-terminal region of mTOR in several cancer types, and one is specifically abundant in kidney cancer, all of which maintain the sensitivity to mTOR inhibition by pharmacological therapies [65].
Moreover, genetic aberrations in components of mTOR complexes are reported to have a close relationship with cancer. RICTOR, a component of mTORC2, was found to be amplified in beast cancer, non-small cell lung cancer (NSCLC), and particularly in squamous cell lung carcinoma (SQCLC), in which RICTOR amplification is significantly related to poor prognosis and short survival [66][67][68]. Overexpression of RICTOR was also observed in gliomas with high Akt activity in nearly 70% of patients and HER2 (human epidermal growth factor receptor-2)-positive breast cancers, leading to Akt hyperactivity and tumor aggravation [67][69].
Except for the above, mTOR signaling hyper-activation can commonly result from mutations of upstream genes including oncogenes and tumor suppressor genes [70]. The PI3K signaling pathway, which is upstream of both mTOR complexes, often has various kinds of mutations of its components in cancer, such as mutation and amplification of Akt and of PIK3CA and amplification of growth factor receptors, Epidermal Growth Factor Receptor (EGFR) and insulin growth factor receptor (IGFR) [71][72][73]. Since PI3K and RAS are two parallel pathways, amplification of growth factor receptors that are upstream of either signal can also result in abnormal signal transduction on both mTOR complexes [74]. Furthermore, loss of functions in tumor suppressor genes, such as PTEN, p53, TSC1/TSC2 and Serine Threonine Kinase 11 (STK11), all contribute to mTOR activation in the pathological state of cancer [75]. PTEN, which is the second most frequently mutated gene after p53 in human cancer, can be downregulated through mutation, methylation, protein instability and intracellular localization [76]. Aberrations in the PTEN genes also influence cancer cells in myeloma, breast cancer and endometrium cancer, which are sensitive to mTOR inhibitors [77][78][79][80]. Inactivation of TSC1 or TSC2, which are negative regulators of mTORC1, is responsible for Tuberous Sclerosis and leads to benign tumor genesis. This also demonstrates that mTORC1 serves as a potent driver of cell proliferation. Mutations of TSC1 and TSC2 are reported in bladder cancer, urothelial carcinoma, clear cell renal carcinoma and well-differentiated pancreatic neuroendocrine tumors [81][82][83]. Actually, mutations in TSC1, TSC2 and mTOR are much less frequent than those in components that are higher upstream in the signaling pathway.
mTOR signaling mainly regulates cell proliferation and metabolism involved in tumor initiation and progression. As reported, at the level of 4E-BP1/eIF-4E, dysregulation of protein synthesis downstream of mTORC1 play a central role in tumorigenesis. eIF-4E promotes the translation of specific pro-oncogenic proteins that regulate cell survival, cell cycle progression, angiogenesis, energy metabolism, and metastasis. Besides, mTOR activation also leads to increased ribosome biogenesis, providing machinery to maintain high levels of cell growth [14]. In cancer cells, metabolism seems to reprogram to sustain the demands of rapid cell growth. mTOR complex is recently depicted as a nutrient sensor in metabolism of cancer, especially on glucose and amino acid, nucleotide, fatty acid and lipid, growth factors and other stresses. Nutrient sensing mainly activates mTORC1 and the metabolic changes in cancer cells sustain mTOC1 activation in turn [44][16][17][84]. In glucose metabolism, mTORC1 can enhance the translation of two key transcription factors, hypoxia inducible factor (HIF)-1α and Myc, which drive expression of a variety of glycolytic enzymes to regulate glycolysis [85][86][87]. mTORC2 can also increase glucose metabolism through its downstream effector AKT [88]. For lipid synthesis, mTORC1 activates the critical transcription factor sterol regulatory element-binding protein 1 (SRE-BP1) driving gene transcription in lipid synthesis via Akt activation and phosphorylation of Lipin1 and S6K1 [89][90]. The increased levels of SRE-BP mRNA and protein are associated with mTORC1 upregulation in human breast cancer tissues [91]. In addition, purine and pyrimidine synthesis, which is significant for cancer cell DNA replication, can also be promoted by mTORC1 via S6K1 phosphorylation [92][93].
Moreover, mTOR is involved in the regulation of autophagy, a process that degrades and recycles cytosolic components in response to a shortage of nutrients and energy. Autophagy is commonly regarded as an inhibition process against tumorigenesis, and blockage of autophagy contributes to cancer initiation [94]. However, some conflicting research results have demonstrated that autophagy may play a dual role in cancer development under specific conditions: for example, it is dependent on different P53 status in pancreatic cancer [95][96][97]. mTORC1 is reported to inactivate UNC-5-like autophagy-activating kinase 1 (ULK1) by phosphorylation resulting in failure to form ULK1-ATG13-FIP200 complex, which is required for autophagy initiation [98][99][100], while mTORC2 can inhibit autophagy indirectly by activating mTORC1. mTORC1 also regulates autophagy at the transcription level by modulating a key transcription factor, Transcription Factor EB (TFEB), for genes in lysosomes and autophagy [101]. Moreover, mTORC1 is likely to affect autophagy through some other ways such as the death-associated protein 1 (DAP1) which suppresses autophagy, WD repeat domain phophoinositide-interacting protein 2 (WIPI2) and a mammalian ortholog of Atg18 [17].
This entry is adapted from the peer-reviewed paper 10.3390/ijms20030755
References
- Bakkenist, C.J.; Kastan, M.B. Initiating cellular stress responses. Cell 2004, 118, 9–17.
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909.
- Conciatori, F.; Ciuffreda, L.; Bazzichetto, C.; Falcone, I.; Pilotto, S.; Bria, E.; Cognetti, F.; Milella, M. mTOR Cross-Talk in Cancer and Potential for Combination Therapy. Cancers 2018, 10, 23.
- Rad, E.; Murray, J.T.; Tee, A.R. Oncogenic Signalling through Mechanistic Target of Rapamycin (mTOR): A Driver of Metabolic Transformation and Cancer Progression. Cancers 2018, 10, 5.
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162.
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168.
- Fonseca, B.D.; Smith, E.M.; Lee, V.H.; MacKintosh, C.; Proud, C.G. PRAS40 is a target for mammalian target of rapamycin complex 1 and is required for signaling downstream of this complex. J. Biol. Chem. 2007, 282, 24514–24524.
- Peterson, T.R.; Laplante, M.; Thoreen, C.C.; Sancak, Y.; Kang, S.A.; Kuehl, W.M.; Gray, N.S.; Sabatini, D.M. DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 2009, 137, 873–886.
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209.
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302.
- Pearce, L.R.; Huang, X.; Boudeau, J.; Pawlowski, R.; Wullschleger, S.; Deak, M.; Ibrahim, A.F.; Gourlay, R.; Magnuson, M.A.; Alessi, D.R. Identification of Protor as a novel Rictor-binding component of mTOR complex-2. Biochem. J. 2007, 405, 513–522.
- Woo, S.Y.; Kim, D.H.; Jun, C.B.; Kim, Y.M.; Haar, E.V.; Lee, S.I.; Hegg, J.W.; Bandhakavi, S.; Griffin, T.J.; Kim, D.H. PRR5, a novel component of mTOR complex 2, regulates platelet-derived growth factor receptor beta expression and signaling. J. Biol. Chem. 2007, 282, 25604–25612.
- Kim, L.C.; Cook, R.S.; Chen, J. mTORC1 and mTORC2 in cancer and the tumor microenvironment. Oncogene 2017, 36, 2191–2201.
- Laplante, M.; Sabatini, D.M. mTOR signaling in growth control and disease. Cell 2012, 149, 274–293.
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484.
- Harachi, M.; Masui, K.; Okamura, Y.; Tsukui, R.; Mischel, P.S.; Shibata, N. mTOR Complexes as a Nutrient Sensor for Driving Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3267.
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in Cancer and Autophagy. Cancers 2018, 10, 18.
- Ben-Sahra, I.; Manning, B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017, 45, 72–82.
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128.
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22.
- Nobes, C.D.; Hall, A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J. Cell Biol. 1999, 144, 1235–1244.
- Zhang, H.; Bajraszewski, N.; Wu, E.; Wang, H.; Moseman, A.P.; Dabora, S.L.; Griffin, J.D.; Kwiatkowski, D.J. PDGFRs are critical for PI3K/Akt activation and negatively regulated by mTOR. J. Clin. Investig. 2007, 117, 730–738.
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657.
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162.
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665.
- Puertollano, R. mTOR and lysosome regulation. F1000Prime Rep. 2014, 6, 52.
- Sancak, Y.; Thoreen, C.C.; Peterson, T.R.; Lindquist, R.A.; Kang, S.A.; Spooner, E.; Carr, S.A.; Sabatini, D.M. PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol. Cell 2007, 25, 903–915.
- Ballif, B.A.; Roux, P.P.; Gerber, S.A.; MacKeigan, J.P.; Blenis, J.; Gygi, S.P. Quantitative phosphorylation profiling of the ERK/p90 ribosomal S6 kinase-signaling cassette and its targets, the tuberous sclerosis tumor suppressors. Proc. Natl. Acad. Sci. USA 2005, 102, 667–672.
- Carriere, A.; Cargnello, M.; Julien, L.A.; Gao, H.; Bonneil, E.; Thibault, P.; Roux, P.P. Oncogenic MAPK signaling stimulates mTORC1 activity by promoting RSK-mediated raptor phosphorylation. Curr. Biol. 2008, 18, 1269–1277.
- Ma, L.; Chen, Z.; Erdjument-Bromage, H.; Tempst, P.; Pandolfi, P.P. Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 2005, 121, 179–193.
- Feng, Z.; Zhang, H.; Levine, A.J.; Jin, S. The coordinate regulation of the p53 and mTOR pathways in cells. Proc. Natl. Acad. Sci. USA 2005, 102, 8204–8209.
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226.
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303.
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501.
- Bonfils, G.; Jaquenoud, M.; Bontron, S.; Ostrowicz, C.; Ungermann, C.; De Virgilio, C. Leucyl-tRNA synthetase controls TORC1 via the EGO complex. Mol. Cell 2012, 46, 105–110.
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA synthetase is an intracellular leucine sensor for the mTORC1-signaling pathway. Cell 2012, 149, 410–424.
- Wang, S.; Tsun, Z.Y.; Wolfson, R.L.; Shen, K.; Wyant, G.A.; Plovanich, M.E.; Yuan, E.D.; Jones, T.D.; Chantranupong, L.; Comb, W.; et al. Metabolism. Lysosomal amino acid transporter SLC38A9 signals arginine sufficiency to mTORC1. Science 2015, 347, 188–194.
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48.
- Ma, X.M.; Blenis, J. Molecular mechanisms of mTOR-mediated translational control. Nat. Rev. Mol. Cell Biol. 2009, 10, 307–318.
- Gingras, A.C.; Kennedy, S.G.; O’Leary, M.A.; Sonenberg, N.; Hay, N. 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt(PKB) signaling pathway. Genes Dev. 1998, 12, 502–513.
- Browne, G.J.; Proud, C.G. A novel mTOR-regulated phosphorylation site in elongation factor 2 kinase modulates the activity of the kinase and its binding to calmodulin. Mol. Cell Biol. 2004, 24, 2986–2997.
- Holz, M.K.; Ballif, B.A.; Gygi, S.P.; Blenis, J. mTOR and S6K1 mediate assembly of the translation preinitiation complex through dynamic protein interchange and ordered phosphorylation events. Cell 2005, 123, 569–580.
- Hsieh, A.C.; Liu, Y.; Edlind, M.P.; Ingolia, N.T.; Janes, M.R.; Sher, A.; Shi, E.Y.; Stumpf, C.R.; Christensen, C.; Bonham, M.J.; et al. The translational landscape of mTOR signalling steers cancer initiation and metastasis. Nature 2012, 485, 55–61.
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371.
- Azpiazu, I.; Saltiel, A.R.; DePaoli-Roach, A.A.; Lawrence, J.C. Regulation of both glycogen synthase and PHAS-I by insulin in rat skeletal muscle involves mitogen-activated protein kinase-independent and rapamycin-sensitive pathways. J. Biol. Chem. 1996, 271, 5033–5039.
- Hudson, C.C.; Liu, M.; Chiang, G.G.; Otterness, D.M.; Loomis, D.C.; Kaper, F.; Giaccia, A.J.; Abraham, R.T. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol. Cell Biol. 2002, 22, 7004–7014.
- Huffman, T.A.; Mothe-Satney, I.; Lawrence, J.C., Jr. Insulin-stimulated phosphorylation of lipin mediated by the mammalian target of rapamycin. Proc. Natl. Acad. Sci. USA 2002, 99, 1047–1052.
- Peterson, R.T.; Desai, B.N.; Hardwick, J.S.; Schreiber, S.L. Protein phosphatase 2A interacts with the 70-kDa S6 kinase and is activated by inhibition of FKBP12-rapamycinassociated protein. Proc. Natl. Acad. Sci. USA 1999, 96, 4438–4442.
- Yokogami, K.; Wakisaka, S.; Avruch, J.; Reeves, S.A. Serine phosphorylation and maximal activation of STAT3 during CNTF signaling is mediated by the rapamycin target mTOR. Curr. Biol. 2000, 10, 47–50.
- Dowling, R.J.; Topisirovic, I.; Alain, T.; Bidinosti, M.; Fonseca, B.D.; Petroulakis, E.; Wang, X.; Larsson, O.; Selvaraj, A.; Liu, Y.; et al. mTORC1-mediated cell proliferation, but not cell growth, controlled by the 4E-BPs. Science 2010, 328, 1172–1176.
- Fingar, D.C.; Salama, S.; Tsou, C.; Harlow, E.; Blenis, J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev. 2002, 16, 1472–1487.
- Humphrey, S.J.; Yang, G.; Yang, P.; Fazakerley, D.J.; Stockli, J.; Yang, J.Y.; James, D.E. Dynamic adipocyte phosphoproteome reveals that Akt directly regulates mTORC2. Cell Metab. 2013, 17, 1009–1020.
- Liu, P.; Gan, W.; Inuzuka, H.; Lazorchak, A.S.; Gao, D.; Arojo, O.; Liu, D.; Wan, L.; Zhai, B.; Yu, Y.; et al. Sin1 phosphorylation impairs mTORC2 complex integrity and inhibits downstream Akt signalling to suppress tumorigenesis. Nat. Cell Biol. 2013, 15, 1340–1350.
- Yang, G.; Murashige, D.S.; Humphrey, S.J.; James, D.E. A Positive Feedback Loop between Akt and mTORC2 via SIN1 Phosphorylation. Cell Rep. 2015, 12, 937–943.
- Zhang, J.; Xu, K.; Liu, P.; Geng, Y.; Wang, B.; Gan, W.; Guo, J.; Wu, F.; Chin, Y.R.; Berrios, C.; et al. Inhibition of Rb Phosphorylation Leads to mTORC2-Mediated Activation of Akt. Mol. Cell 2016, 62, 929–942.
- Willems, L.; Tamburini, J.; Chapuis, N.; Lacombe, C.; Mayeux, P.; Bouscary, D. PI3K and mTOR signaling pathways in cancer: New data on targeted therapies. Curr. Oncol. Rep. 2012, 14, 129–138.
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322.
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205.
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villen, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326.
- Bakker, W.J.; Harris, I.S.; Mak, T.W. FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol. Cell 2007, 28, 941–953.
- Weiler, M.; Blaes, J.; Pusch, S.; Sahm, F.; Czabanka, M.; Luger, S.; Bunse, L.; Solecki, G.; Eichwald, V.; Jugold, M.; et al. mTOR target NDRG1 confers MGMT-dependent resistance to alkylating chemotherapy. Proc. Natl. Acad. Sci. USA 2014, 111, 409–414.
- Gan, X.; Wang, J.; Wang, C.; Sommer, E.; Kozasa, T.; Srinivasula, S.; Alessi, D.; Offermanns, S.; Simon, M.I.; Wu, D. PRR5L degradation promotes mTORC2-mediated PKC-delta phosphorylation and cell migration downstream of Galpha12. Nat. Cell Biol. 2012, 14, 686–696.
- Thomanetz, V.; Angliker, N.; Cloetta, D.; Lustenberger, R.M.; Schweighauser, M.; Oliveri, F.; Suzuki, N.; Ruegg, M.A. Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J. Cell Biol. 2013, 201, 293–308.
- Fruman, D.A.; Rommel, C. PI3K and cancer: Lessons, challenges and opportunities. Nat. Rev. Drug Discov. 2014, 13, 140–156.
- Grabiner, B.C.; Nardi, V.; Birsoy, K.; Possemato, R.; Shen, K.; Sinha, S.; Jordan, A.; Beck, A.H.; Sabatini, D.M. A diverse array of cancer-associated MTOR mutations are hyperactivating and can predict rapamycin sensitivity. Cancer Discov. 2014, 4, 554–563.
- Cheng, H.; Zou, Y.; Ross, J.S.; Wang, K.; Liu, X.; Halmos, B.; Ali, S.M.; Liu, H.; Verma, A.; Montagna, C.; et al. RICTOR Amplification Defines a Novel Subset of Patients with Lung Cancer Who May Benefit from Treatment with mTORC1/2 Inhibitors. Cancer Discov. 2015, 5, 1262–1270.
- Morrison Joly, M.; Hicks, D.J.; Jones, B.; Sanchez, V.; Estrada, M.V.; Young, C.; Williams, M.; Rexer, B.N.; Sarbassov dos, D.; Muller, W.J.; et al. Rictor/mTORC2 Drives Progression and Therapeutic Resistance of HER2-Amplified Breast Cancers. Cancer Res. 2016, 76, 4752–4764.
- Balko, J.M.; Giltnane, J.M.; Wang, K.; Schwarz, L.J.; Young, C.D.; Cook, R.S.; Owens, P.; Sanders, M.E.; Kuba, M.G.; Sanchez, V.; et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014, 4, 232–245.
- Masri, J.; Bernath, A.; Martin, J.; Jo, O.D.; Vartanian, R.; Funk, A.; Gera, J. mTORC2 activity is elevated in gliomas and promotes growth and cell motility via overexpression of rictor. Cancer Res. 2007, 67, 11712–11720.
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832 e823.
- Guertin, D.A.; Sabatini, D.M. Defining the role of mTOR in cancer. Cancer Cell 2007, 12, 9–22.
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug Discov. 2009, 8, 627–644.
- Guertin, D.A.; Stevens, D.M.; Saitoh, M.; Kinkel, S.; Crosby, K.; Sheen, J.H.; Mullholland, D.J.; Magnuson, M.A.; Wu, H.; Sabatini, D.M. mTOR complex 2 is required for the development of prostate cancer induced by Pten loss in mice. Cancer Cell 2009, 15, 148–159.
- Mayer, I.A.; Arteaga, C.L. The PI3K/AKT Pathway as a Target for Cancer Treatment. Annu. Rev. Med. 2016, 67, 11–28.
- Zoncu, R.; Efeyan, A.; Sabatini, D.M. mTOR: From growth signal integration to cancer, diabetes and ageing. Nat. Rev. Mol. Cell Biol. 2011, 12, 21–35.
- Milella, M.; Falcone, I.; Conciatori, F.; Cesta Incani, U.; Del Curatolo, A.; Inzerilli, N.; Nuzzo, C.M.; Vaccaro, V.; Vari, S.; Cognetti, F.; et al. PTEN: Multiple Functions in Human Malignant Tumors. Front. Oncol. 2015, 5, 24.
- Shi, Y.; Gera, J.; Hu, L.; Hsu, J.H.; Bookstein, R.; Li, W.; Lichtenstein, A. Enhanced sensitivity of multiple myeloma cells containing PTEN mutations to CCI-779. Cancer Res. 2002, 62, 5027–5034.
- DeGraffenried, L.A.; Fulcher, L.; Friedrichs, W.E.; Grunwald, V.; Ray, R.B.; Hidalgo, M. Reduced PTEN expression in breast cancer cells confers susceptibility to inhibitors of the PI3 kinase/Akt pathway. Ann. Oncol. 2004, 15, 1510–1516.
- Milam, M.R.; Celestino, J.; Wu, W.; Broaddus, R.R.; Schmeler, K.M.; Slomovitz, B.M.; Soliman, P.T.; Gershenson, D.M.; Wang, H.; Ellenson, L.H.; et al. Reduced progression of endometrial hyperplasia with oral mTOR inhibition in the Pten heterozygote murine model. Am. J. Obstet. Gynecol. 2007, 196, 247-e1.
- Pulito, C.; Mori, F.; Sacconi, A.; Goeman, F.; Ferraiuolo, M.; Pasanisi, P.; Campagnoli, C.; Berrino, F.; Fanciulli, M.; Ford, R.J.; et al. Metformin-induced ablation of microRNA 21-5p releases Sestrin-1 and CAB39L antitumoral activities. Cell Discov. 2017, 3, 17022.
- Platt, F.M.; Hurst, C.D.; Taylor, C.F.; Gregory, W.M.; Harnden, P.; Knowles, M.A. Spectrum of phosphatidylinositol 3-kinase pathway gene alterations in bladder cancer. Clin. Cancer Res. 2009, 15, 6008–6017.
- Sjodahl, G.; Lauss, M.; Gudjonsson, S.; Liedberg, F.; Hallden, C.; Chebil, G.; Mansson, W.; Hoglund, M.; Lindgren, D. A systematic study of gene mutations in urothelial carcinoma; inactivating mutations in TSC2 and PIK3R1. PLoS ONE 2011, 6, e18583.
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroendocrine tumors. Science 2011, 331, 1199–1203.
- Mossmann, D.; Park, S.; Hall, M.N. mTOR signalling and cellular metabolism are mutual determinants in cancer. Nat. Rev. Cancer 2018, 18, 744–757.
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183.
- Majumder, P.K.; Febbo, P.G.; Bikoff, R.; Berger, R.; Xue, Q.; McMahon, L.M.; Manola, J.; Brugarolas, J.; McDonnell, T.J.; Golub, T.R.; et al. mTOR inhibition reverses Akt-dependent prostate intraepithelial neoplasia through regulation of apoptotic and HIF-1-dependent pathways. Nat. Med. 2004, 10, 594–601.
- Gordan, J.D.; Thompson, C.B.; Simon, M.C. HIF and c-Myc: Sibling rivals for control of cancer cell metabolism and proliferation. Cancer Cell 2007, 12, 108–113.
- Elstrom, R.L.; Bauer, D.E.; Buzzai, M.; Karnauskas, R.; Harris, M.H.; Plas, D.R.; Zhuang, H.; Cinalli, R.M.; Alavi, A.; Rudin, C.M.; et al. Akt stimulates aerobic glycolysis in cancer cells. Cancer Res. 2004, 64, 3892–3899.
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420.
- Porstmann, T.; Santos, C.R.; Griffiths, B.; Cully, M.; Wu, M.; Leevers, S.; Griffiths, J.R.; Chung, Y.L.; Schulze, A. SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab. 2008, 8, 224–236.
- Ricoult, S.J.; Yecies, J.L.; Ben-Sahra, I.; Manning, B.D. Oncogenic PI3K and K-Ras stimulate de novo lipid synthesis through mTORC1 and SREBP. Oncogene 2016, 35, 1250–1260.
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328.
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733.
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46.
- Rosenfeldt, M.T.; O’Prey, J.; Morton, J.P.; Nixon, C.; MacKay, G.; Mrowinska, A.; Au, A.; Rai, T.S.; Zheng, L.; Ridgway, R.; et al. p53 status determines the role of autophagy in pancreatic tumour development. Nature 2013, 504, 296–300.
- Iacobuzio-Donahue, C.A.; Herman, J.M. Autophagy, p53, and pancreatic cancer. N. Engl. J. Med. 2014, 370, 1352–1353.
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316.
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32.
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991.
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003.
- Settembre, C.; Fraldi, A.; Medina, D.L.; Ballabio, A. Signals from the lysosome: A control centre for cellular clearance and energy metabolism. Nat. Rev. Mol. Cell Biol. 2013, 14, 283–296.
This entry is offline, you can click here to edit this entry!