Obesity is caused by prolonged energy surplus. Current anti-obesity medications are mostly centralized around the energy input part of the energy balance equation by increasing satiety and reducing appetite. Our gastrointestinal tract is a key organ for regulation of food intake and supplies a tremendous number of circulating signals that modulate the activity of appetite-regulating areas of the brain by either direct interaction or through the vagus nerve. Intestinally derived messengers are manifold and include absorbed nutrients, microbial metabolites, gut hormones and other enterokines, collectively comprising a fine-tuned signalling system to the brain. After a meal, nutrients directly interact with appetite-inhibiting areas of the brain and induce satiety. However, overall feeding behaviour also depends on secretion of gut hormones produced by highly specialized and sensitive enteroendocrine cells. Moreover, circulating microbial metabolites and their interactions with enteroendocrine cells further contribute to the regulation of feeding patterns. Current therapies exploiting the appetite-regulating properties of the gut are based on chemically modified versions of the gut hormone, glucagon-like peptide-1 (GLP-1) or on inhibitors of the primary GLP-1 inactivating enzyme, dipeptidyl peptidase-4 (DPP-4). The effectiveness of these approaches shows that that the gut is a promising target for therapeutic interventions to achieve significant weigh loss.
- enteroendocrine cells
- enterokines
- gut microbiota
1. Introduction
Intestinally derived signals act on receptors on vagus nerves and neurons in appetite-controlling areas of the brain. In obesity, satiety signalling is inhibited, resulting in excessive food intake. Although a vast number of studies have been carried out to dissect the mechanisms that prevent the normal satiety in individuals living with obesity, the identity of the driving factor(s) is lacking. Most studies on obesity pathogenesis suggest the main cause is central dysregulation of feeding behaviour and lack of hunger suppression in the brain. Impaired gut hormone secretion and dysbiosis are often associated with obesity, but it is unclear whether this perturbed signalling has a causative role or whether its long-term perturbed signalling persists after weight loss and/or weight regain. Nevertheless, the signals derived from the intestine in the process of nutrient processing and uptake can be extremely powerful to supress the appetite. For example, the recent success of the GLP-1-based analogues for weight loss provides clear evidence of the potential for utilizing the intestinally-derived signalling pathway for obesity treatment. However, the intestine itself as a drug target can provide many more ways of modulating the energy intake.
Nutrient signalling is the primary signal initiating satiety at several levels and each one of them offers an opportunity to influence the satiety circuits—such as modulating the absorption rate or gut hormone secretion. Keeping in mind that the intestine is the largest inner organ in the body and contributes with 20–35% of whole-body energy expenditure [1], the diet-induced energy expenditure component is important for energy homeostasis. The intestinal epithelial lining is heavily vascularized and equipped with an underlying network of lymphatic capillaries which not only allow nutrient absorption but also interaction with immune cells. Recent studies show that these interactions are tightly connected to the satiety signalling. In addition, metabolites from symbiotic microorganisms, contributing to digestion, form signalling pathways to various systems of the human body, such as liver, immune system, and brain, helping to maintain energy homeostasis. As impaired regulation of food intake is the main driving force in obesity, focusing on origins of the natural satiety signalling and developing new ways of boosting it may provide us with better and safer ways of controlling the appetite.
2. Intestinal Epithelium and Signalling Properties of Nutrients
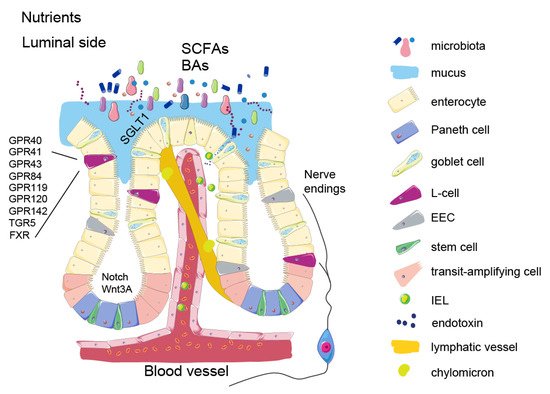
3. Enteroendocrine Cells
4. Microbial Metabolites
5. Enterokines and Gut-Liver Axis
6. Intestinal Barrier Function
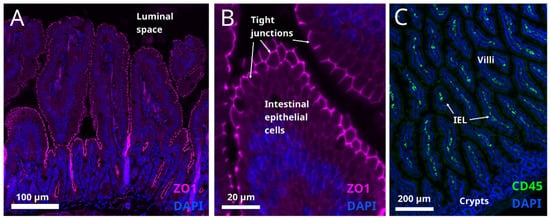
This entry is adapted from the peer-reviewed paper 10.3390/metabo12010039
References
- Van Der Schoor, S.R.D.; Reeds, P.J.; Stoll, B.; Henry, J.F.; Rosenberger, J.R.; Burrin, D.G.; Van Goudoever, J.B. The High Metabolic Cost of a Functional Gut. Gastroenterology 2002, 123, 1931–1940.
- Clevers, H. The Intestinal Crypt, a Prototype Stem Cell Compartment. Cell 2013, 154, 274–284.
- Gehart, H.; Clevers, H. Tales from the Crypt: New Insights into Intestinal Stem Cells. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 19–34.
- Mah, A.T.; Van Landeghem, L.; Gavin, H.E.; Magness, S.T.; Lund, P.K. Impact of Diet-Induced Obesity on Intestinal Stem Cells: Hyperproliferation but Impaired Intrinsic Function That Requires Insulin/IGF1. Endocrinology 2014, 155, 3302–3314.
- Aliluev, A.; Tritschler, S.; Sterr, M.; Oppenländer, L.; Hinterdobler, J.; Greisle, T.; Irmler, M.; Beckers, J.; Sun, N.; Walch, A.; et al. Diet-Induced Alteration of Intestinal Stem Cell Function Underlies Obesity and Prediabetes in Mice. Nat. Metab. 2021, 3, 1202–1216.
- Pourvali, K.; Monji, H. Obesity and Intestinal Stem Cell Susceptibility to Carcinogenesis. Nutr. Metab. 2021, 18, 37.
- Tysoe, O. PPAR Mediates Intestinal Stem Cell Tumorigenesis. Nat. Rev. Endocrinol. 2021, 17, 514.
- Dailey, M.J. Nutrient-Induced Intestinal Adaption and Its Effect in Obesity. Physiol. Behav. 2014, 136, 74–78.
- Yang, W.; Bancroft, L.; Nicholas, C.; Lozonschi, I.; Augenlicht, L.H. Targeted Inactivation of P27kip1 Is Sufficient for Large and Small Intestinal Tumorigenesis in the Mouse, Which Can Be Augmented by a Western-Style High-Risk Diet. Cancer Res. 2003, 63, 4990–4996.
- Gerbe, F.; Jay, P. Intestinal Tuft Cells: Epithelial Sentinels Linking Luminal Cues to the Immune System. Mucosal Immunol. 2016, 9, 1353–1359.
- Furness, J.B.; Rivera, L.R.; Cho, H.-J.; Bravo, D.M.; Callaghan, B. The Gut as a Sensory Organ. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 729–740.
- Lund, M.L.; Egerod, K.L.; Engelstoft, M.S.; Dmytriyeva, O.; Theodorsson, E.; Patel, B.A.; Schwartz, T.W. Enterochromaffin 5-HT Cells-A Major Target for GLP-1 and Gut Microbial Metabolites. Mol. Metab. 2018, 11, 70–83.
- Kato, S. Role of serotonin 5-HT3 receptors in intestinal inflammation. Biol. Pharm. Bull. 2013, 36, 1406–1409.
- Oh, C.M.; Park, S.; Kim, H. Serotonin as a New Therapeutic Target for Diabetes Mellitus and Obesity. Diabetes Metab. J. 2016, 40, 89–98.
- Binetti, J.; Bertran, L.; Riesco, D.; Aguilar, C.; Martínez, S.; Sabench, F.; Porras, J.A.; Camaron, J.; Castillo, D.D.; Richart, C.; et al. Deregulated Serotonin Pathway in Women with Morbid Obesity and NAFLD. Life 2020, 10, 245.
- Carey, A.L.; Kingwell, B.A. Reducing peripheral serotonin turns up the heat in brown fat. Nat. Med. 2015, 21, 114–116.
- Singh, R.K.; Chang, H.-W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of Diet on the Gut Microbiome and Implications for Human Health. J. Transl. Med. 2017, 15, 73.
- Tolhurst, G.; Heffron, H.; Lam, Y.S.; Parker, H.E.; Habib, A.M.; Diakogiannaki, E.; Cameron, J.; Grosse, J.; Reimann, F.; Gribble, F.M. Short-Chain Fatty Acids Stimulate Glucagon-like Peptide-1 Secretion via the G-Protein-Coupled Receptor FFAR2. Diabetes 2012, 61, 364–371.
- Nøhr, M.K.; Pedersen, M.H.; Gille, A.; Egerod, K.L.; Engelstoft, M.S.; Husted, A.S.; Sichlau, R.M.; Grunddal, K.V.; Poulsen, S.S.; Han, S.; et al. GPR41/FFAR3 and GPR43/FFAR2 as Cosensors for Short-Chain Fatty Acids in Enteroendocrine Cells vs. FFAR3 in Enteric Neurons and FFAR2 in Enteric Leukocytes. Endocrinology 2013, 154, 3552–3564.
- Fellows, R.; Denizot, J.; Stellato, C.; Cuomo, A.; Jain, P.; Stoyanova, E.; Balázsi, S.; Hajnády, Z.; Liebert, A.; Kazakevych, J.; et al. Microbiota Derived Short Chain Fatty Acids Promote Histone Crotonylation in the Colon through Histone Deacetylases. Nat. Commun. 2018, 9, 105.
- Larraufie, P.; Martin-Gallausiaux, C.; Lapaque, N.; Dore, J.; Gribble, F.M.; Reimann, F.; Blottiere, H.M. SCFAs Strongly Stimulate PYY Production in Human Enteroendocrine Cells. Sci. Rep. 2018, 8, 74.
- Borgeraas, H.; Johnson, L.K.; Skattebu, J.; Hertel, J.K.; Hjelmesaeth, J. Effects of probiotics on body weight, body mass index, fat mass and fat percentage in subjects with overweight or obesity: A systematic review and meta-analysis of randomized controlled trials. Obes. Rev. 2018, 19, 219–232.
- Koutnikova, H.; Genser, B.; Monteiro-Sepulveda, M.; Faurie, J.M.; Rizkalla, S.; Schrezenmeir, J.; Clément, K. Impact of bacterial probiotics on obesity, diabetes and non-alcoholic fatty liver disease related variables: A systematic review and meta-analysis of randomised controlled trials. BMJ Open 2019, 9, e017995.
- Ilmonen, J.; Isolauri, E.; Poussa, T.; Laitinen, K. Impact of dietary counselling and probiotic intervention on maternal anthropometric measurements during and after pregnancy: A randomized placebo-controlled trial. Clin. Nutr. 2011, 30, 156–164.
- Depommier, C.; Everard, A.; Druart, C.; Plovier, H.; Van Hul, M.; Vieira-Silva, S.; Falony, G.; Raes, J.; Maiter, D.; Delzenne, N.M.; et al. Supplementation with Akkermansia Muciniphila in Overweight and Obese Human Volunteers: A Proof-of-Concept Exploratory Study. Nat. Med. 2019, 25, 1096–1103.
- Fon Tacer, K.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research Resource: Comprehensive Expression Atlas of the Fibroblast Growth Factor System in Adult Mouse. Mol. Endocrinol. 2010, 24, 2050–2064.
- Marcelin, G.; Jo, Y.-H.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.-M.; Blouet, C.; Chang, J.K.; Chua, S. Central Action of FGF19 Reduces Hypothalamic AGRP/NPY Neuron Activity and Improves Glucose Metabolism. Mol. Metab. 2014, 3, 19–28.
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro-O, M. Tissue-Specific Expression of BetaKlotho and Fibroblast Growth Factor (FGF) Receptor Isoforms Determines Metabolic Activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695.
- Holt, J.A.; Luo, G.; Billin, A.N.; Bisi, J.; McNeill, Y.Y.; Kozarsky, K.F.; Donahee, M.; Wang, D.Y.; Mansfield, T.A.; Kliewer, S.A.; et al. Definition of a Novel Growth Factor-Dependent Signal Cascade for the Suppression of Bile Acid Biosynthesis. Genes Dev. 2003, 17, 1581–1591.
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast Growth Factor 15 Functions as an Enterohepatic Signal to Regulate Bile Acid Homeostasis. Cell Metab. 2005, 2, 217–225.
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 Regulates Hepatic Glucose Metabolism by Inhibiting the CREB-PGC-1α Pathway. Cell Metab. 2011, 13, 729–738.
- Song, K.-H.; Li, T.; Owsley, E.; Strom, S.; Chiang, J.Y.L. Bile Acids Activate Fibroblast Growth Factor 19 Signaling in Human Hepatocytes to Inhibit Cholesterol 7alpha-Hydroxylase Gene Expression. Hepatology 2009, 49, 297–305.
- Kim, Y.-C.; Seok, S.; Zhang, Y.; Ma, J.; Kong, B.; Guo, G.; Kemper, B.; Kemper, J.K. Intestinal FGF15/19 Physiologically Repress Hepatic Lipogenesis in the Late Fed-State by Activating SHP and DNMT3A. Nat. Commun. 2020, 11, 5969.
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a Postprandial, Insulin-Independent Activator of Hepatic Protein and Glycogen Synthesis. Science 2011, 331, 1621–1624.
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast Growth Factor 19 Increases Metabolic Rate and Reverses Dietary and Leptin-Deficient Diabetes. Endocrinology 2004, 145, 2594–2603.
- Morton, G.J.; Matsen, M.E.; Bracy, D.P.; Meek, T.H.; Nguyen, H.T.; Stefanovski, D.; Bergman, R.N.; Wasserman, D.H.; Schwartz, M.W. FGF19 Action in the Brain Induces Insulin-Independent Glucose Lowering. J. Clin. Investig. 2013, 123, 4799–4808.
- Ryan, K.K.; Kohli, R.; Gutierrez-Aguilar, R.; Gaitonde, S.G.; Woods, S.C.; Seeley, R.J. Fibroblast Growth Factor-19 Action in the Brain Reduces Food Intake and Body Weight and Improves Glucose Tolerance in Male Rats. Endocrinology 2013, 154, 9–15.
- Lan, T.; Morgan, D.A.; Rahmouni, K.; Sonoda, J.; Fu, X.; Burgess, S.C.; Holland, W.L.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19, FGF21, and an FGFR1/β-Klotho-Activating Antibody Act on the Nervous System to Regulate Body Weight and Glycemia. Cell Metab. 2017, 26, 709–718.e3.
- Wellen, K.E.; Hotamisligil, G.S. Inflammation, Stress, and Diabetes. J. Clin. Investig. 2005, 115, 1111–1119.
- Pendyala, S.; Walker, J.M.; Holt, P.R. A High-Fat Diet Is Associated with Endotoxemia That Originates from the Gut. Gastroenterology 2012, 142, 1100–1101.e2.
- Cani, P.D.; Osto, M.; Geurts, L.; Everard, A. Involvement of Gut Microbiota in the Development of Low-Grade Inflammation and Type 2 Diabetes Associated with Obesity. Gut Microbes 2012, 3, 279–288.