Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Endocrinology & Metabolism
DICER1 protein is a member of the ribonuclease (RNAse) III family with a key role in the biogenesis of microRNAs (miRNA) and in microRNA processing, potentially affecting gene regulation at the post-transcriptional level.
- DICER1
- Thyroid
- Enzyme
- Gene
- Endocrine pathology
- Thyroid nodules
- Thyroid pathology
1. “The Gene DICER1” and the “Enzyme DICER”
The DICER1 gene is located on chromosome 14q32.13. It is composed of 27 exons and encodes a 1922-aminoacid protein with a molecular weight of approximately 200 kDa [6]. DICER1 encodes a multidomain enzyme that belongs to the RNase III family. DICER protein domains orderly locate from the N- to the C-terminus and include the following domains: Helicase (Hel1, Hel2i and Hel2), DUF283, Platform, PAZ (Piwi/Argonaut/Zwille), Connector helix, RNase IIIa and IIIb and dsRNA-binding domain (dsRBD) (Figure 1). Structurally, DICER has three rigid regions: RNase III, Platform-PAZ and helicase [6,15].
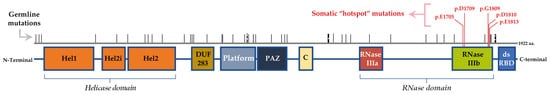
Figure 1. Schematic representation of the hDICER protein domains and the location of hotspot somatic mutations. hDICER1 is composed by the following domains, from N- to C-terminus of the protein: helicase domain (Hel1, Hel2i and Hel2), DUF283, platform, Piwi-Argonaute-Zwille (PAZ), connector helix (C), RNase IIIa, RNase IIIb and double-stranded RNA-binding domain (dsRBD). The hotspot somatic mutations for DICER1 gene in thyroid lesions locate in the RNase IIIb domain, while the germline mutations can be found along all the gene.
In a very simplistic description, the helicase domain allows the opening of the double-stranded RNA structures of the precursor microRNAs (pre-miRNAs) that are then cut by the RNase IIIa and IIIb domains in the 3p and 5p strand, respectively. The other domains are crucial to support these processing reactions from pre-miRNA to miRNA, namely the Platform, PAZ and Connector helix domains, which are important for the recognition, and the dsRBD domain needed for the binding of DICER1 to pre-miRNAs [13,16].
In the canonical path, the DICER multi-domain enzyme plays a crucial role in the biogenesis of the small RNAs, namely miRNAs (Figure 2) [15,17,18]. This is a cytoplasmic stepwise process that is incorporated into a specific pathway involving the large family of Argonaute (AGO) proteins and large multiprotein complexes termed RNA-induced silencing complexes (RISC) [17,18]. These processes guide for the sequence-specific silencing of genes through mRNA degradation, translational repression and heterochromatin formation. Beyond the canonical role of DICER via small RNA biogenesis in the cytoplasm, accumulated evidence suggests a non-canonical, non-endonuclease role of DICER in the nucleus. These processes are briefly summarized in Figure 2.
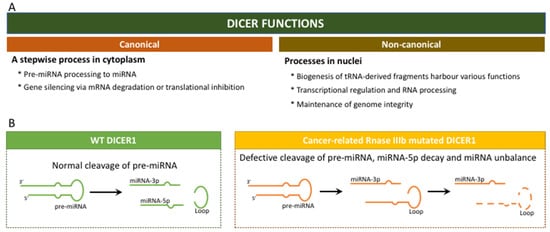
Figure 2. DICER1 functions: (A) Canonical and non-canonical cellular functions; (B) Canonical functions of DICER1 in a context of wild-type DICER1 and in a context of mutated DICER1, in hotspot RNase IIIb domain.
2. Mutations/Alterations of DICER1
Normal development and tumorigenesis of thyroid gland share many common pathways involved in cell proliferation and differentiation. The importance of miRNAs, as well as the role of DICER1 in processing precursor miRNA to mature ones, was evident in the maintenance of the thyroid tissue homeostasis, as well as in its involvement in tumorigenesis [19,20].
As will be discussed below, germline mutations in the DICER1 gene occur in any part of the gene, while somatic mutations are particularly frequent in the ribonuclease (ribonuclease IIIb) domain (Figure 1). The germline mutations are mostly inactivating mutations that cause DICER1 loss of function (LOF) and, as consequence, the downregulation of miRNA levels [9,20]. Mutations may be seen either in individuals in the context of DICER1 syndromic cases or in predisposed carriers. The LOF germline mutations can occur via deletion of the entire locus of the gene, as in- and out-of-frame intragenic deletions for one or more exons and also as somatic mosaicism. As supported by animal model studies, DICER1 does not act as the classical tumour suppressor genes or oncogenes [21]. To be able to promote tumorigenesis, the partial loss of DICER1 is required, indicating its role as a haploinsufficient tumour suppressor [2,9]. It was estimated that germline pathogenic variants of DICER1 may lead to LOF occurring in a ratio of 1:10,600 in the population [22]. Intriguingly, thyroid lesions seen in both germline and somatic mutations of the DICER1 gene were not found to be associated with other canonical thyroid genetic events in paediatric patients, such as BRAF, RAS or TERTp mutations [12].
3. Thyroid Neoplasm in the Context of DICER1 Syndromic and Non-Syndromic Mutations
The predisposition to the manifestation of thyroid lesions is increased when the individual presents germline syndromic mutations [23]. However, if the germline alteration is present in one allele only, the chance of developing a lesion in the thyroid gland is low. A second event is usually necessary. The second event can be the presence of a DICER1 somatic mutation in the second allele. In the context of DICER1 syndrome with previous clinical manifestations, screening for DICER1 somatic mutations is standard when thyroid lesions are identified in the individuals. Unlike what was previously reported [12], the second event can be unrelated to additional alterations in DICER1; it can be related with the presence of genetic alterations in other genes that increase the risk of the occurrence of thyroid lesions (benign and/or malignant).
Initially, DICER1-related thyroid manifestations were thought to be restricted to MNG or FA, but with the increase in case reports, large cohorts and population data-base studies on both DICER1 germline and somatic testing, a phenotypically diverse spectrum of thyroid entities, ranging from the most benign manifestations to the most aggressive tumours, was revealed [2]. MNG is frequently associated with DICER1 syndrome, which can be diagnosed from early ages until the fourth decade of life [8]. Despite being less frequent than MNG, differentiated thyroid tumours are also seen in DICER1 syndrome, with a higher incidence in the first decades of life [24]. Individuals who harbour a germline DICER1 alteration have a 16-fold increased risk of developing a thyroid tumour, and the incidence of MNG is higher in female than in male carriers [8]. While MNG is not a life-threatening lesion, differentiated thyroid carcinoma (DTC) or poorly differentiated thyroid carcinoma (PDTC) diagnosis, especially in young people, is associated with some risk of mortality [25]. For this reason, when a DICER1 syndrome or DICER1 alterations in an individual are known, a closer surveillance for thyroid lesions (as well as those of other organs) must follow, since early diagnosis can improve patient follow-up, and therefore lead to a more favourable prognosis. The diagnosis of any thyroid lesion, especially in early life, even in the absence of a familial history, should also be considered as a warning of the possibility of alterations in DICER1. So, molecular testing of DICER1 may be beneficial to the patient and, ultimately, to their relatives.
Recently, Stewart et al. studied the risk of the appearance of various types of neoplasms associated with DICER1 syndrome [26]. There is a range of ages with a high risk of certain types of neoplasms associated with DICER1 syndrome. For instance, the risk of PPB is high in the first years of life (up to 6 years), while the risk for Sertoli–Leydig cell tumours (SLCT) and other neoplasms (which include those of the thyroid) is distributed from early ages to 20 years of age (and there is still a slight increase up to around the age 40) [26].
Thyroid gland lesions are the paediatric syndromic forms that have been reported the most in the context of DICER1 syndrome [23], but thyroid tumours are also found in the range of 20–40 years of age [26]. Thyroid lesions are less common in the oldest age groups, i.e., more than 40 years. Nonetheless, they are still represented. Two issues that are pertinent to the aforementioned age distribution profile deserves discussion. Firstly, the lesions may have been temporarily diagnosed long after their initial development and, due to this, the association of the age at diagnosis with the presence of DICER1 mutations may be somewhat biased. Secondly, the anticipated knowledge of the presence of DICER1 germline mutations in families/individuals in most studies leads to a closer clinical surveillance, supporting an early detection of lesions.
Two studies highlighted the results in paediatric thyroid tumours [11,27]. Chernock et al. studied six cases of paediatric PDTC and found that the presence of DICER1 somatic mutations in those aggressive TC is relatively common (four out of six, all of them with a hotspot in the RNAse IIIB domain), whereas germline mutations are less common (one out of six, affecting one region of splicing). These cases suggest that, in the presence of relatively more aggressive tumours at younger ages, the evaluation for the presence of mutations in the DICER1 gene could be beneficial, but further studies are needed in larger series [11]. Bae et al. studied a series of 41 paediatric follicular thyroid tumours (adenoma and carcinoma), whose patients had no previous history of DICER1 syndrome-associated lesions. Using NGS, the authors found that DICER1 somatic mutations were more common (9 out of 41 cases) than mutations in thyroid-related genes, such as NRAS (6 out of 41 cases), HRAS, PTEN, TSHR, RET and others. In that study, DICER1 and NRAS alterations were found to be mutually exclusive [27].
Knowing that DICER1 somatic mutations, although not very common, can occur and represent a tumorigenic event, it would be advisable to include DICER1 hotspot mutations analysis in the panels of genes studied for the molecular profiling of thyroid lesions. The study of mutations for DICER1 in commercial applications is already performed, namely in ThyroSeq v3 [28]. In a large study carried out by Chong et al., the authors found the presence of DICER1 hotspot mutations in 1.4% of the cohort studied (214 out of 14,993 FNAs). In the same study, Chong et al. found that, although not absent in DICER1 mutated lesions—changes in other genes that are related with thyroid tumorigenesis (RAS, BRAF, RET, TSHR, EIF1AX and TP53, among others) are less common in lesions that present DICER1 hotspot mutations [28]. Taking this fact into consideration, and the evidence of more recent studies demonstrating that DICER1 mutations are not restricted to benign thyroid lesions and may contribute biologically to more aggressive thyroid tumours, the molecular testing for DICER1 could be recommended to patients whose thyroid lesions are diagnosed before the age of 40, when typically one would not expect to see genetic alterations in the most common thyroid cancer-related genes as often and where an individualized treatment and follow-up approach is required the most. Similarly, a more thorough molecular study on poorly differentiated TC and some variants of PTC may be of interest, given their apparent higher relative prevalence in DICER1-mutated patients. In such cases, when there is a clinical indication to look for the presence of hotspot mutations in the DICER1 gene, this can be done by PCR followed by Sanger sequencing. These are well-established and robust techniques that provide a good cost–benefit ratio for the screening of hotspot mutations.
When searching for mutations or variants in the entire DICER1 gene, the benefit of using different techniques must be considered. Since the gene is very large, it can be performed either by the laborious amplification of several amplicons by PCR/Sanger sequencing or by the use of more advanced techniques, such as NGS. While in cases of suspected DICER1 syndrome the second option seems to be the most obvious, given the lack of a predominant region for the appearance of germline alterations, when searching for somatic alterations the use of NGS should only be considered if hotspot mutations were not detected. Even so, the search for somatic mutations beyond the RNase IIIb hotspot needs additional supportive studies to ascertain its clinical relevance. Most studies reported hotspot mutations in thyroid lesions, mainly because of their screening focused on this region of the gene. Studies on TCGA database did not find other somatic mutations either; however, a recent report using the MSK-IMPACT database described additional ones [2,13,29].
4. DICER1 Somatic Hotspot Mutations and the Free Pass to Thyroid Malignancy
As mentioned above, DICER1-related tumours typically and most frequently harbour an “RNase IIIb hotspot” somatic mutation and less frequently LOH. The tumour-specific RNase IIIb hotspot mutations are missense mutations that normally occur in one of the five codons that encode for the protein catalytic domain residues (p.E1705, p.D1709, p.G1809, p.D1810 and p.E1813). These DICER1 hotspot mutations result in altered activity of DICER1 protein in microRNA processing and lead to a rapid and improper 5p miRNA cleavage and consequent degradation. Because of the 5p strand miRNAs loss, there is a consistent change in the microRNA landscape, as well as in the messenger RNA (mRNA) profiles mediated by the RISC complex. Somatic RNase hotspot mutations typically partner with a second alteration to promote tumorigenesis. They can couple in three fashions: somatic with germline, somatic with mosaic and somatic with somatic [9]. Although there is a remarkable miRNA imbalance/deregulation in the presence of somatic DICER1 mutation, this does not mean that it will always lead to malignancy. Indeed, MNG is the most frequent thyroid entity in DICER1 syndromic cases, but malignant entities ranging from the most innocent to the aggressive ones were more often associated with somatic mutations [37]. Oliver-Petit and al. [7], reported a series of eight families referred for childhood-onset of MNG or DICER1-related tumours with a familial history of MNG. The authors found that DICER1 somatic pathogenic mutations were present in both benign and malignant thyroid nodules, suggesting that the thyroid carcinogenesis pathway may be somehow unique in the background of DICER1-syndromic cases. The frequency of DICER1 mutations is higher in the background of LOF germline mutations, and RNase IIIb hotspot mutations were found to be more frequently associated with the presence of malignant tumours [9]. Noteworthily, more recent studies have documented a higher prevalence of DICER1 variants in an adolescent-onset PTC group [12,33]. In the study of Lee at el., although the number of FTC was far too low to draw any conclusion, all the cases of somatic mutations were observed in FTC only [12]. Based on this, the group draw the attention to the possible utility of somatic DICER1 testing in a young age group, particularly in the presence of a family history of MNG, thyroid surgery or associated embryonal tumour. Wasserman et al. noted the absence of thyroid autoimmunity lesions and local or distant metastasis in this group of PTCs, whereas Lee et al. attributed the favourable course of the cases to the predominance of miFTC cases in their series [12,33]. Nonetheless, these two studies pointed out the low risk of the malignant lesions in paediatric PTC and FTC groups. More recently, somatic mutations of DICER1 were also found to be related with PDTC and teratocarcinomas of the thyroid [6]. On this point, Agaimy et al. have reported two cases of DICER1 sporadic malignant teratoid thyroid tumours while thoroughly reviewing the clinicopathological and molecular characteristics of six additional cases previously reported [38]. In their report, the authors point out the highly aggressive course of this entity in comparison with the low malignant potential of other organ blastomas and proposed the term “thyroblastoma” for the disease. These cases tend to present at higher ages, without any family or personal history of other neoplasms, and have somatic mutations of DICER1, mostly a hotspot, thus supporting the concept of a DICER1 sporadic, non-syndromic form of this neoplasm and the need to differentiate it from the classical teratomas or carcinosarcomas [38].
Of those RNase IIIb hotspot mutations, there is a particular group of mutations called “mosaicism for RNase IIIb domain hotspot mutations” in DICER1 syndrome patients that deserves mention. According to Brenneman et al. and based on the study of 124 patients from the International PPB Registry (IPPBR), this group should be distinguished from the germline and mosaic LOF mutations due to their peculiar clinical characteristics—the disease tends to occur much earlier in life, and multisite disease is frequent. Shultz et al. [39] emphasized the importance of this subgroup, underlying two points: (a) First, whereas in the presence of a germline mutation (usually LOF) there is a need of a second hotspot mutation in the RNAse IIIb domain (limited to a very small target site(s)), in the presence of mosaicism hotspot mutations there is “only” a need for an additional alteration that causes LOF (whose probability of occurring is hundreds of times more likely); (b) Second, there is also the chance that the allele combination of hotspot and wild type together could be tumorigenic on its own. Therefore, a group of authors and working groups from Bakhuizen et al. suggested that the patients with mosaicism for hotspot should be under intensive and long-term surveillance. Today, the individuals with somatic mosaicism are known to show increased penetrance of DICER1 syndrome, including an earlier onset, higher number of disease foci and wider range of phenotypes [40].
This entry is adapted from the peer-reviewed paper 10.3390/jmp3010001
This entry is offline, you can click here to edit this entry!