G protein-coupled receptors (GPCRs) are a superfamily of proteins classically described as monomeric transmembrane (TM) receptors. However, increasing evidence indicates that many GPCRs form higher-order assemblies made up of monomers pertaining to identical (homo) or to various (hetero) receptors. The formation and structure of these oligomers, their physiological role and possible therapeutic applications raise a variety of issues that are currently being actively explored. In this context, synthetic peptides derived from TM domains stand out as powerful tools that can be predictably targeted to disrupt GPCR oligomers, especially at the interface level, eventually impairing their action.
- peptide therapeutics
- transmembrane peptides
- GPCR oligomers
- non-natural amino acids
- cyclic peptides
- retro-enantio
1. Introduction
2. GPCR Oligomers
3. Synthetic TM Peptides as Tools for GPCR Complex Exploration
|
GPCR Complex |
TMs Involved in Dimerization |
Synthetic TM Disruptor Peptide |
In Vitro/In Vivo Assays Performed |
Patho-Physiological Implication |
Ref. |
|---|---|---|---|---|---|
|
A2AR-D2R |
TM4/5 interface |
A2AR TM5 |
|
Cocaine use |
[53] |
|
APJR-OX1R |
TM4/5 interface |
APJ TM4, TM5 |
|
- |
[60] |
|
APJR homodimer |
TM1, TM2, TM3, TM4 |
TM1, TM2, TM3, TM4 |
|
- |
[61] |
|
A2AR-CB1R |
TM 5/6 interface |
CB1R TM5 TM6 A2AR TM5 TM6 |
|
Glutamate release |
[59] |
|
A1R-A2AR |
TM 5/6 interface |
A2AR TM4, TM5, TM6 A1R TM5 and TM6 |
|
Neurodegeneration Neuroinflammation |
[62] |
|
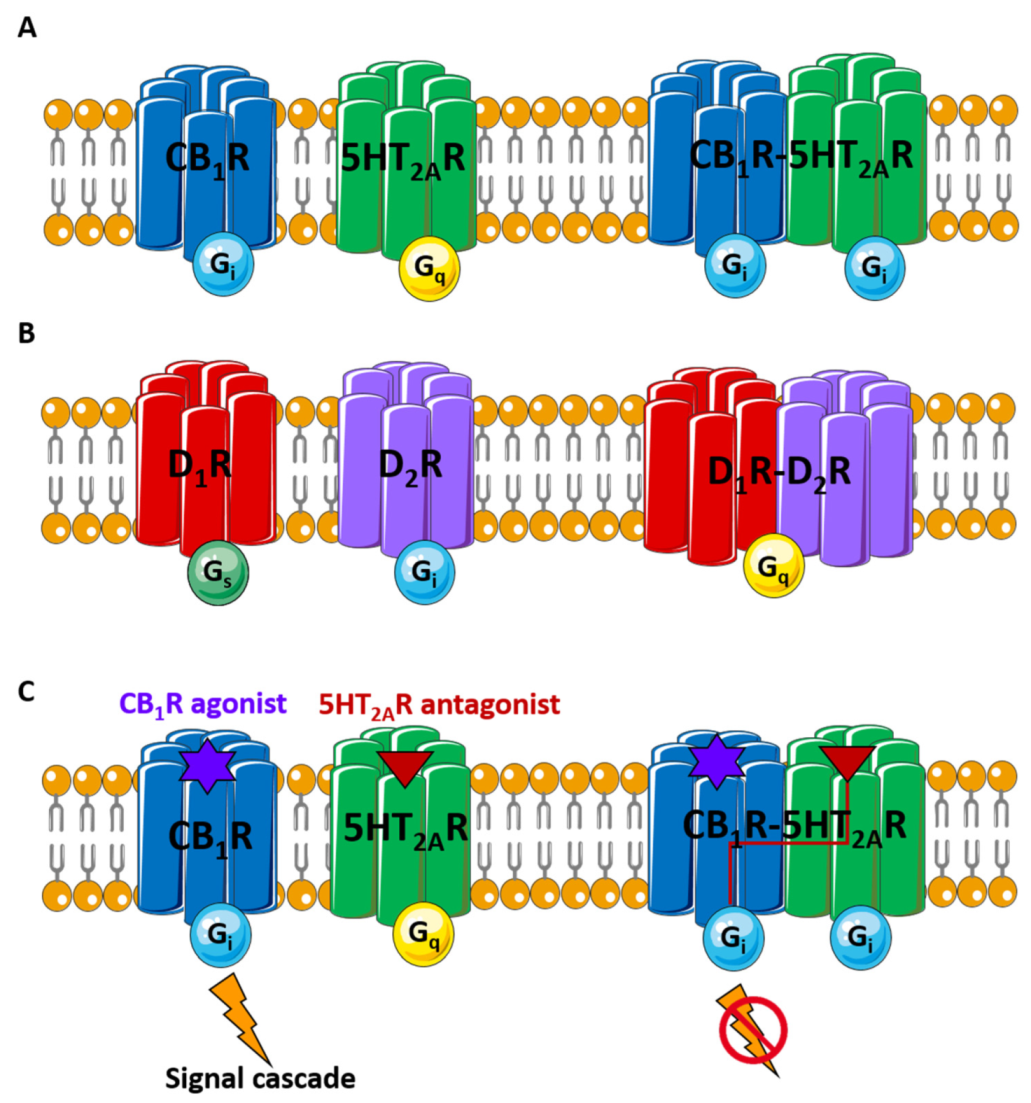
CB1R-5HT2AR |
TM 5/6 interface |
CB1R TM5, TM6 |
|
Cognitive impairment |
[40] |
|
M3R homodimer |
TM1, TM5, TM7 |
TM1-TM5-TM7 |
|
- |
[63] |
|
CCKR homodimer |
TM6 |
TM6 |
|
- |
[64] |
|
CCR5 homodimer |
TM1, TM2, TM4 |
TM1, TM4 |
|
- |
[65] |
|
RhoR homodimer |
TM1,TM2, TM4, TM5, H8 |
TM1, TM2, TM4, TM5 |
|
Phototransduction |
[66] |
|
β2AR homodimer |
TM1, TM5, TM6, H8 |
TM6 |
|
- |
[17] |
|
SCTR |
TM4 |
TM4 |
|
Liver diseases |
[55] |
|
AT1aR-SCTR |
TM1/2 interface TM4/4 interface |
AT1aR TM1, TM4 SCTR TM2, TM4 |
|
Hyperosmolality-induced drinking |
[54] |
|
FZD6 homodimer |
TM4, TM5 |
TM4, TM5 |
|
Cancer and neurologic disorders |
[67] |
|
MOR-DOR |
MOR TM1 |
MOR TM1 |
|
Morphine tolerance |
[68] |
Abbreviations: 5HT2AR, serotonin receptor type 2 A; A1R, adenosine receptor type 1; A2AR, adenosine receptor type 2A; APJR, apelin receptor; AT1aR, angiotensin receptor type 1a; BiFC, bimolecular fluorescence complementation; BRET, bioluminescence resonance energy transfer; cAMP, cyclic adenosine monophosphate; CB1R, cannabinoid receptor type 1; CCKR, cholecystokinin receptor; CCR5, chemokine receptor type 5; CODA-RET, complemented donor-acceptor resonance energy transfer; Co-IP, co-immunoprecipitation; D2R, dopamine receptor type 2; DMR, dynamic mass redistribution; DOR, δ-opioid receptor; FCCS, fluorescence cross-correlation spectroscopy; FRAP, fluorescence recovery after photobleaching; FRET, fluorescence resonance energy transfer; FZD6R, Frizzled-6 receptor; M3R, muscarinic acetylcholine receptor type 3; MOR, μ-opioid receptors; NORT, novel object recognition test; OX1R, orexin receptor type 1; PLA, proximity ligation assay; RhoR, rhodopsin receptor; SCTR, secretin receptor; TIRF, total internal reflection fluorescence; β2AR, adrenergic receptor type β2.
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics14010161
References
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650.
- Lefkowitz, R.J. Historical review: A brief history and personal retrospective of seven-transmembrane receptors. Trends Pharmacol. Sci. 2004, 25, 413–422.
- Limbird, L.E.; Meyts, P.D.; Lefkowitz, R.J. Beta-adrenergic receptors: Evidence for negative cooperativity. Biochem. Biophys. Res. Commun. 1975, 64, 1160–1168.
- Wettschureck, N.; Offermanns, S. Mammalian G proteins and their cell type specific functions. Physiol. Rev. 2005, 85, 1159–1204.
- Attwood, T.K.; Findlay, J.B. Fingerprinting G-protein-coupled receptors. Protein Eng. 1994, 7, 195–203.
- Tuteja, N. Signaling through G protein coupled receptors. Plant Signal. Behav. 2009, 4, 942–947.
- Wacker, D.; Stevens, R.C.; Roth, B.L. How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427.
- Ferré, S.; Bonaventura, J.; Tomasi, D.; Navarro, G.; Moreno, E.; Cortés, A.; Lluís, C.; Casadó, V.; Volkow, N.D. Allosteric mechanisms within the adenosine A2A-dopamine D2 receptor heterotetramer. Neuropharmacology 2016, 104, 154–160.
- Cook, J.L. G protein-coupled receptors as disease targets: Emerging paradigms. Ochsner J. 2010, 10, 2–7.
- Sriram, K.; Insel, P.A. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol. Pharmacol. 2018, 93, 251–258.
- Jacobson, K.A. New paradigms in GPCR drug discovery. Biochem. Pharmacol. 2015, 98, 541–555.
- Nieto Gutierrez, A.; McDonald, P.H. GPCRs: Emerging anti-cancer drug targets. Cell. Signal. 2018, 41, 65–74.
- Hauser, A.S.; Chavali, S.; Masuho, I.; Jahn, L.J.; Martemyanov, K.A.; Gloriam, D.E.; Babu, M.M. Pharmacogenomics of GPCR Drug Targets. Cell 2018, 172, 41–54.e19.
- Glukhova, A.; Draper-Joyce, C.J.; Sunahara, R.K.; Christopoulos, A.; Wootten, D.; Sexton, P.M. Rules of Engagement: GPCRs and G Proteins. ACS Pharmacol. Transl. Sci. 2018, 1, 73–83.
- Milligan, G.; Ward, R.J.; Marsango, S. GPCR homo-oligomerization. Curr. Opin. Cell Biol. 2019, 57, 40–47.
- Fuxe, K.; Borroto-Escuela, D.O.; Marcellino, D.; Romero-Fernandez, W.; Frankowska, M.; Guidolin, D.; Filip, M.; Ferraro, L.; Woods, A.S.; Tarakanov, A.; et al. GPCR heteromers and their allosteric receptor-receptor interactions. Curr. Med. Chem. 2012, 19, 356–363.
- Hebert, T.E.; Moffett, S.; Morello, J.P.; Loisel, T.P.; Bichet, D.G.; Barret, C.; Bouvier, M. A peptide derived from a beta2-adrenergic receptor transmembrane domain inhibits both receptor dimerization and activation. J. Biol. Chem. 1996, 271, 16384–16392.
- Fredriksson, R.; Lagerström, M.C.; Lundin, L.-G.; Schiöth, H.B. The G-protein-coupled receptors in the human genome form five main families. Phylogenetic analysis, paralogon groups, and fingerprints. Mol. Pharmacol. 2003, 63, 1256–1272.
- Li, X.; Zhou, M.; Huang, W.; Yang, H. N-glycosylation of the β(2) adrenergic receptor regulates receptor function by modulating dimerization. FEBS J. 2017, 284, 2004–2018.
- Bagher, A.M.; Young, A.P.; Laprairie, R.B.; Toguri, J.T.; Kelly, M.E.M.; Denovan-Wright, E.M. Heteromer formation between cannabinoid type 1 and dopamine type 2 receptors is altered by combination cannabinoid and antipsychotic treatments. J. Neurosci. Res. 2020, 98, 2496–2509.
- Ferré, S.; Ciruela, F. Functional and Neuroprotective Role of Striatal Adenosine A(2A) Receptor Heterotetramers. J. Caffeine Adenosine Res. 2019, 9, 89–97.
- Bono, F.; Mutti, V.; Fiorentini, C.; Missale, C. Dopamine D3 Receptor Heteromerization: Implications for Neuroplasticity and Neuroprotection. Biomolecules 2020, 10, 1016.
- Borroto-Escuela, D.O.; Tarakanov, A.O.; Brito, I.; Fuxe, K. Glutamate heteroreceptor complexes in the brain. Pharmacol. Rep. 2018, 70, 936–950.
- Bontempi, L.; Savoia, P.; Bono, F.; Fiorentini, C.; Missale, C. Dopamine D3 and acetylcholine nicotinic receptor heteromerization in midbrain dopamine neurons: Relevance for neuroplasticity. Eur. Neuropsychopharmacol. J. Eur. Coll. Neuropsychopharmacol. 2017, 27, 313–324.
- Guitart, X.; Navarro, G.; Moreno, E.; Yano, H.; Cai, N.-S.; Sánchez-Soto, M.; Kumar-Barodia, S.; Naidu, Y.T.; Mallol, J.; Cortés, A.; et al. Functional selectivity of allosteric interactions within G protein-coupled receptor oligomers: The dopamine D1-D3 receptor heterotetramer. Mol. Pharmacol. 2014, 86, 417–429.
- Cai, N.-S.; Quiroz, C.; Bonaventura, J.; Bonifazi, A.; Cole, T.O.; Purks, J.; Billing, A.S.; Massey, E.; Wagner, M.; Wish, E.D.; et al. Opioid-galanin receptor heteromers mediate the dopaminergic effects of opioids. J. Clin. Investig. 2019, 129, 2730–2744.
- Wang, J.; Hua, T.; Liu, Z.-J. Structural features of activated GPCR signaling complexes. Curr. Opin. Struct. Biol. 2020, 63, 82–89.
- Borroto-Escuela, D.O.; Ferraro, L.; Narvaez, M.; Tanganelli, S.; Beggiato, S.; Liu, F.; Rivera, A.; Fuxe, K. Multiple Adenosine-Dopamine (A2A-D2 Like) Heteroreceptor Complexes in the Brain and Their Role in Schizophrenia. Cells 2020, 9, 1077.
- Perreault, M.L.; Hasbi, A.; O’Dowd, B.F.; George, S.R. Heteromeric dopamine receptor signaling complexes: Emerging neurobiology and disease relevance. Neuropsychopharmacology 2014, 39, 156–168.
- Farran, B. An update on the physiological and therapeutic relevance of GPCR oligomers. Pharmacol. Res. 2017, 117, 303–327.
- Jordan, B.A.; Devi, L.A. G-protein-coupled receptor heterodimerization modulates receptor function. Nature 1999, 399, 697–700.
- Xue, L.; Sun, Q.; Zhao, H.; Rovira, X.; Gai, S.; He, Q.; Pin, J.-P.; Liu, J.; Rondard, P. Rearrangement of the transmembrane domain interfaces associated with the activation of a GPCR hetero-oligomer. Nat. Commun. 2019, 10, 2765.
- Kasai, R.S.; Ito, S.V.; Awane, R.M.; Fujiwara, T.K.; Kusumi, A. The Class-A GPCR Dopamine D2 Receptor Forms Transient Dimers Stabilized by Agonists: Detection by Single-Molecule Tracking. Cell Biochem. Biophys. 2018, 76, 29–37.
- Tabor, A.; Weisenburger, S.; Banerjee, A.; Purkayastha, N.; Kaindl, J.M.; Hübner, H.; Wei, L.; Grömer, T.W.; Kornhuber, J.; Tschammer, N.; et al. Visualization and ligand-induced modulation of dopamine receptor dimerization at the single molecule level. Sci. Rep. 2016, 6, 33233.
- Aslanoglou, D.; Alvarez-Curto, E.; Marsango, S.; Milligan, G. Distinct Agonist Regulation of Muscarinic Acetylcholine M2-M3 Heteromers and Their Corresponding Homomers. J. Biol. Chem. 2015, 290, 14785–14796.
- Gao, Y.; Westfield, G.; Erickson, J.W.; Cerione, R.A.; Skiniotis, G.; Ramachandran, S. Isolation and structure-function characterization of a signaling-active rhodopsin-G protein complex. J. Biol. Chem. 2017, 292, 14280–14289.
- Navarro, G.; Cordomi, A.; Zelman-Femiak, M.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Perez-Benito, L.; Cortes, A.; Casado, V.; Mallol, J.; et al. Quaternary structure of a G-protein-coupled receptor heterotetramer in complex with Gi and Gs. BMC Biol. 2016, 14, 26.
- Cordomí, A.; Navarro, G.; Aymerich, M.S.; Franco, R. Structures for G-Protein-Coupled Receptor Tetramers in Complex with G Proteins. Trends Biochem. Sci. 2015, 40, 548–551.
- Deganutti, G.; Salmaso, V.; Moro, S. Could Adenosine Recognize its Receptors with a Stoichiometry Other than 1:1? Mol. Inform. 2018, 37, e1800009.
- Vinals, X.; Moreno, E.; Lanfumey, L.; Cordomi, A.; Pastor, A.; de La Torre, R.; Gasperini, P.; Navarro, G.; Howell, L.A.; Pardo, L.; et al. Cognitive Impairment Induced by Delta9-tetrahydrocannabinol Occurs through Heteromers between Cannabinoid CB1 and Serotonin 5-HT2A Receptors. PLoS Biol. 2015, 13, e1002194.
- Rashid, A.J.; So, C.H.; Kong, M.M.C.; Furtak, T.; El-Ghundi, M.; Cheng, R.; O’Dowd, B.F.; George, S.R. D1-D2 dopamine receptor heterooligomers with unique pharmacology are coupled to rapid activation of Gq/11 in the striatum. Proc. Natl. Acad. Sci. USA 2007, 104, 654–659.
- Bellot, M.; Galandrin, S.; Boularan, C.; Matthies, H.J.; Despas, F.; Denis, C.; Javitch, J.; Mazères, S.; Sanni, S.J.; Pons, V.; et al. Dual agonist occupancy of AT1-R-α2C-AR heterodimers results in atypical Gs-PKA signaling. Nat. Chem. Biol. 2015, 11, 271–279.
- Baba, K.; Benleulmi-Chaachoua, A.; Journé, A.-S.; Kamal, M.; Guillaume, J.-L.; Dussaud, S.; Gbahou, F.; Yettou, K.; Liu, C.; Contreras-Alcantara, S.; et al. Heteromeric MT1/MT2 melatonin receptors modulate photoreceptor function. Sci. Signal. 2013, 6, ra89.
- Smith, N.J.; Milligan, G. Allostery at G protein-coupled receptor homo- and heteromers: Uncharted pharmacological landscapes. Pharmacol. Rev. 2010, 62, 701–725.
- León-Navarro, D.A.; Albasanz, J.L.; Martín, M. Functional Cross-Talk between Adenosine and Metabotropic Glutamate Receptors. Curr. Neuropharmacol. 2019, 17, 422–437.
- Galvez, T.; Duthey, B.; Kniazeff, J.; Blahos, J.; Rovelli, G.; Bettler, B.; Prézeau, L.; Pin, J.P. Allosteric interactions between GB1 and GB2 subunits are required for optimal GABA(B) receptor function. EMBO J. 2001, 20, 2152–2159.
- Gomes, I.; Jordan, B.A.; Gupta, A.; Trapaidze, N.; Nagy, V.; Devi, L.A. Heterodimerization of mu and delta opioid receptors: A role in opiate synergy. J. Neurosci. 2000, 20, RC110.
- Rocheville, M.; Lange, D.C.; Kumar, U.; Patel, S.C.; Patel, R.C.; Patel, Y.C. Receptors for dopamine and somatostatin: Formation of hetero-oligomers with enhanced functional activity. Science 2000, 288, 154–157.
- Franco, R.; Ferre, S.; Agnati, L.; Torvinen, M.; Gines, S.; Hillion, J.; Casado, V.; Lledo, P.-M.; Zoli, M.; Lluis, C.; et al. Evidence for Adenosine/Dopamine Receptor Interactions: Indications for Heteromerization. Neuropsychopharmacology 2000, 23, S50–S59.
- Navarro, G.; Quiroz, C.; Moreno-Delgado, D.; Sierakowiak, A.; McDowell, K.; Moreno, E.; Rea, W.; Cai, N.-S.; Aguinaga, D.; Howell, L.A.; et al. Orexin-corticotropin-releasing factor receptor heteromers in the ventral tegmental area as targets for cocaine. J. Neurosci. 2015, 35, 6639–6653.
- Martínez-Pinilla, E.; Rodríguez-Pérez, A.I.; Navarro, G.; Aguinaga, D.; Moreno, E.; Lanciego, J.L.; Labandeira-García, J.L.; Franco, R. Dopamine D2 and angiotensin II type 1 receptors form functional heteromers in rat striatum. Biochem. Pharmacol. 2015, 96, 131–142.
- Young, B.M.; Nguyen, E.; Chedrawe, M.A.J.; Rainey, J.K.; Dupré, D.J. Differential Contribution of Transmembrane Domains IV, V, VI, and VII to Human Angiotensin II Type 1 Receptor Homomer Formation. J. Biol. Chem. 2017, 292, 3341–3350.
- Borroto-Escuela, D.O.; Wydra, K.; Li, X.; Rodriguez, D.; Carlsson, J.; Jastrzębska, J.; Filip, M.; Fuxe, K. Disruption of A2AR-D2R Heteroreceptor Complexes After A2AR Transmembrane 5 Peptide Administration Enhances Cocaine Self-Administration in Rats. Mol. Neurobiol. 2018, 55, 7038–7048.
- Lee, L.T.O.; Ng, S.Y.L.; Chu, J.Y.S.; Sekar, R.; Harikumar, K.G.; Miller, L.J.; Chow, B.K.C. Transmembrane peptides as unique tools to demonstrate the in vivo action of a cross-class GPCR heterocomplex. FASEB J. 2014, 28, 2632–2644.
- Harikumar, K.G.; Pinon, D.I.; Miller, L.J. Transmembrane segment IV contributes a functionally important interface for oligomerization of the Class II G protein-coupled secretin receptor. J. Biol. Chem. 2007, 282, 30363–30372.
- Guidolin, D.; Marcoli, M.; Tortorella, C.; Maura, G.; Agnati, L.F. Receptor-Receptor Interactions as a Widespread Phenomenon: Novel Targets for Drug Development? Front. Endocrinol. 2019, 10, 53.
- Møller, T.C.; Hottin, J.; Clerté, C.; Zwier, J.M.; Durroux, T.; Rondard, P.; Prézeau, L.; Royer, C.A.; Pin, J.-P.; Margeat, E.; et al. Oligomerization of a G protein-coupled receptor in neurons controlled by its structural dynamics. Sci. Rep. 2018, 8, 10414.
- Guidolin, D.; Marcoli, M.; Tortorella, C.; Maura, G.; Agnati, L.F. G protein-coupled receptor-receptor interactions give integrative dynamics to intercellular communication. Rev. Neurosci. 2018, 29, 703–726.
- Köfalvi, A.; Moreno, E.; Cordomí, A.; Cai, N.-S.; Fernández-Dueñas, V.; Ferreira, S.G.; Guixà-González, R.; Sánchez-Soto, M.; Yano, H.; Casadó-Anguera, V.; et al. Control of glutamate release by complexes of adenosine and cannabinoid receptors. BMC Biol. 2020, 18, 9.
- Wan, L.; Xu, F.; Liu, C.; Ji, B.; Zhang, R.; Wang, P.; Wu, F.; Pan, Y.; Yang, C.; Wang, C.; et al. Transmembrane peptide 4 and 5 of APJ are essential for its heterodimerization with OX1R. Biochem. Biophys. Res. Commun. 2020, 521, 408–413.
- Cai, X.; Bai, B.; Zhang, R.; Wang, C.; Chen, J. Apelin receptor homodimer-oligomers revealed by single-molecule imaging and novel G protein-dependent signaling. Sci. Rep. 2017, 7, 40335.
- Navarro, G.; Cordomí, A.; Brugarolas, M.; Moreno, E.; Aguinaga, D.; Pérez-Benito, L.; Ferre, S.; Cortés, A.; Casadó, V.; Mallol, J.; et al. Cross-communication between Gi and Gs in a G-protein-coupled receptor heterotetramer guided by a receptor C-terminal domain. BMC Biol. 2018, 16, 24.
- McMillin, S.M.; Heusel, M.; Liu, T.; Costanzi, S.; Wess, J. Structural basis of M3 muscarinic receptor dimer/oligomer formation. J. Biol. Chem. 2011, 286, 28584–28598.
- Harikumar, K.G.; Dong, M.; Cheng, Z.; Pinon, D.I.; Lybrand, T.P.; Miller, L.J. Transmembrane Segment Peptides Can Disrupt Cholecystokinin Receptor Oligomerization without affecting Receptor Function. Biochemistry 2006, 45, 14706–14716.
- Hernanz-Falcón, P.; Rodríguez-Frade, J.M.; Serrano, A.; Juan, D.; del Sol, A.; Soriano, S.F.; Roncal, F.; Gómez, L.; Valencia, A.; Martínez-A, C.; et al. Identification of amino acid residues crucial for chemokine receptor dimerization. Nat. Immunol. 2004, 5, 216–223.
- Jastrzebska, B.; Chen, Y.; Orban, T.; Jin, H.; Hofmann, L.; Palczewski, K. Disruption of Rhodopsin Dimerization with Synthetic Peptides Targeting an Interaction Interface. J. Biol. Chem. 2015, 290, 25728–25744.
- Petersen, J.; Wright, S.C.; Rodríguez, D.; Matricon, P.; Lahav, N.; Vromen, A.; Friedler, A.; Strömqvist, J.; Wennmalm, S.; Carlsson, J.; et al. Agonist-induced dimer dissociation as a macromolecular step in G protein-coupled receptor signaling. Nat. Commun. 2017, 8, 226.
- He, S.-Q.; Zhang, Z.-N.; Guan, J.-S.; Liu, H.-R.; Zhao, B.; Wang, H.-B.; Li, Q.; Yang, H.; Luo, J.; Li, Z.-Y.; et al. Facilitation of mu-opioid receptor activity by preventing delta-opioid receptor-mediated codegradation. Neuron 2011, 69, 120–131.