Obesity is caused by prolonged energy surplus. Current anti-obesity medications are mostly centralized around the energy input part of the energy balance equation by increasing satiety and reducing appetite. Our gastrointestinal tract is a key organ for regulation of food intake and supplies a tremendous number of circulating signals that modulate the activity of appetite-regulating areas of the brain by either direct interaction or through the vagus nerve. Intestinally derived messengers are manifold and include absorbed nutrients, microbial metabolites, gut hormones and other enterokines, collectively comprising a fine-tuned signalling system to the brain. After a meal, nutrients directly interact with appetite-inhibiting areas of the brain and induce satiety. However, overall feeding behaviour also depends on secretion of gut hormones produced by highly specialized and sensitive enteroendocrine cells. Moreover, circulating microbial metabolites and their interactions with enteroendocrine cells further contribute to the regulation of feeding patterns. Current therapies exploiting the appetite-regulating properties of the gut are based on chemically modified versions of the gut hormone, glucagon-like peptide-1 (GLP-1) or on inhibitors of the primary GLP-1 inactivating enzyme, dipeptidyl peptidase-4 (DPP-4). The effectiveness of these approaches shows that that the gut is a promising target for therapeutic interventions to achieve significant weigh loss.
- enteroendocrine cells
- enterokines
- gut microbiota
1. Introduction
Intestinally derived signals act on receptors on vagus nerves and neurons in appetite-controlling areas of the brain. In obesity, satiety signalling is inhibited, resulting in excessive food intake. Although a vast number of studies have been carried out to dissect the mechanisms that prevent the normal satiety in individuals living with obesity, the identity of the driving factor(s) is lacking. Most studies on obesity pathogenesis suggest the main cause is central dysregulation of feeding behaviour and lack of hunger suppression in the brain. Impaired gut hormone secretion and dysbiosis are often associated with obesity, but it is unclear whether this perturbed signalling has a causative role or whether its long-term perturbed signalling persists after weight loss and/or weight regain. Nevertheless, the signals derived from the intestine in the process of nutrient processing and uptake can be extremely powerful to supress the appetite. For example, the recent success of the GLP-1-based analogues for weight loss provides clear evidence of the potential for utilizing the intestinally-derived signalling pathway for obesity treatment. However, the intestine itself as a drug target can provide many more ways of modulating the energy intake.
Nutrient signalling is the primary signal initiating satiety at several levels and each one of them offers an opportunity to influence the satiety circuits—such as modulating the absorption rate or gut hormone secretion. Keeping in mind that the intestine is the largest inner organ in the body and contributes with 20–35% of whole-body energy expenditure [1], the diet-induced energy expenditure component is important for energy homeostasis. The intestinal epithelial lining is heavily vascularized and equipped with an underlying network of lymphatic capillaries which not only allow nutrient absorption but also interaction with immune cells. Recent studies show that these interactions are tightly connected to the satiety signalling. In addition, metabolites from symbiotic microorganisms, contributing to digestion, form signalling pathways to various systems of the human body, such as liver, immune system, and brain, helping to maintain energy homeostasis. As impaired regulation of food intake is the main driving force in obesity, focusing on origins of the natural satiety signalling and developing new ways of boosting it may provide us with better and safer ways of controlling the appetite.
2. Intestinal Epithelium and Signalling Properties of Nutrients
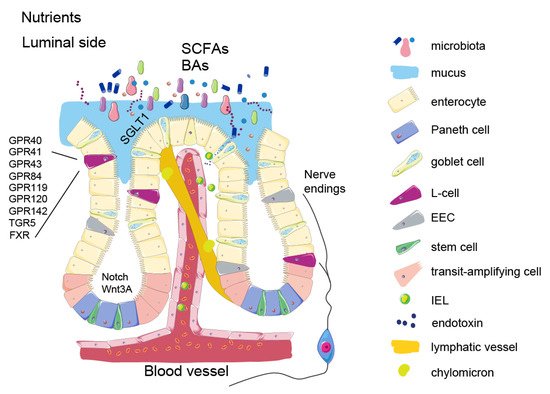
3. Enteroendocrine Cells
4. Microbial Metabolites
5. Enterokines and Gut-Liver Axis
6. Intestinal Barrier Function
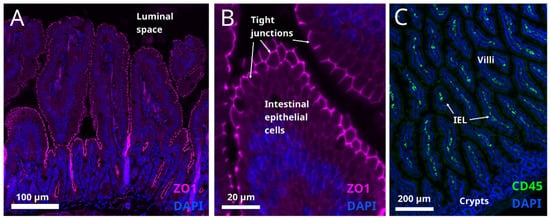
This entry is adapted from the peer-reviewed paper 10.3390/metabo12010039