In the framework of space flight, the risk of radiation carcinogenesis is considered a “red” risk due to the high likelihood of occurrence as well as the high potential impact on the quality of life in terms of disease-free survival after space missions. The cyclic AMP response element-binding protein (CREB) is overexpressed both in haematological malignancies and solid tumours and its expression and function are modulated following irradiation. The CREB protein is a transcription factor and member of the CREB/activating transcription factor (ATF) family. As such, it has an essential role in a wide range of cell processes, including cell survival, proliferation, and differentiation. Among the CREB-related nuclear transcription factors, NF-κB and p53 have a relevant role in cell response to ionising radiation. Their expression and function can decide the fate of the cell by choosing between death or survival.
- CREB/ATF
- NF-κB
- radioresistance
- ionising radiation
- galactic cosmic rays
- radiotherapy
- human cancer
- space flight
1. Introduction
2. CREB Family Members and Related Transcription Factors in Radiotherapy of Solid Tumours and Leukaemia
2.1. Cell Responses to Ionising Radiation
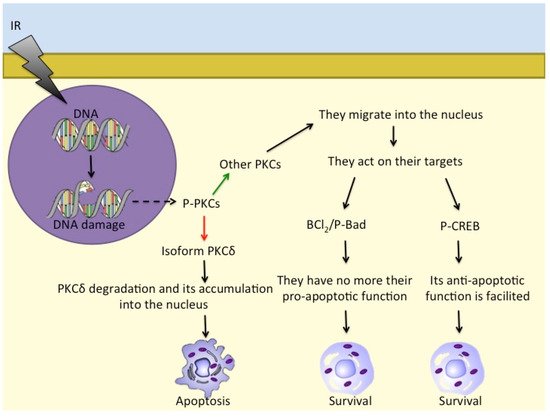
2.2. CREB and Other Factors in Radioresistance and Radiosensitivity
2.3. CREB and Related Transcription Factors as Possible Targets in the Treatment of Tumours
This entry is adapted from the peer-reviewed paper 10.3390/life11121437
References
- El-Jaby, S.; Lewis, B.J.; Tomi, L. A commentary on the impact of modelling results to inform mission planning and shield design. Life Sci. Sp. Res. 2020, 25, 148–150.
- Li, M.; Gonon, G.; Buonanno, M.; Autsavapromporn, N.; De Toledo, S.M.; Pain, D.; Azzam, E.I. Health Risks of Space Exploration: Targeted and Nontargeted Oxidative Injury by High-Charge and High-Energy Particles. Antioxid. Redox Signal. 2014, 20, 1501.
- Romero, E.; Francisco, D. The NASA human system risk mitigation process for space exploration. Acta Astronaut. 2020, 175, 606–615.
- Sridharan, D.M.; Asaithamby, A.; Bailey, S.M.; Costes, S.V.; Doetsch, P.W.; Dynan, W.S.; Kronenberg, A.; Rithidech, K.N.; Saha, J.; Snijders, A.M.; et al. Understanding cancer development processes after HZE-particle exposure: Roles of ROS, DNA damage repair and inflammation. Radiat. Res. 2015, 183, 1–26.
- Prasad, B.; Grimm, D.; Strauch, S.M.; Erzinger, G.S.; Corydon, T.J.; Lebert, M.; Magnusson, N.E.; Infanger, M.; Richter, P.; Krüger, M. Influence of Microgravity on Apoptosis in Cells, Tissues, and Other Systems In Vivo and In Vitro. Int. J. Mol. Sci. 2020, 21, 9373.
- Simonsen, L.C.; Slaba, T.C.; Guida, P.; Rusek, A. NASA’s first ground-based Galactic Cosmic Ray Simulator: Enabling a new era in space radiobiology research. PLoS Biol. 2020, 18, e3000669.
- Barcellos-Hoff, M.H.; Blakely, E.A.; Burma, S.; Fornace, A.J.; Gerson, S.; Hlatky, L.; Kirsch, D.G.; Luderer, U.; Shay, J.; Wang, Y.; et al. Concepts and challenges in cancer risk prediction for the space radiation environment. Life Sci. Sp. Res. 2015, 6, 92–103.
- Montminy, M. Transcriptionl regulation by ciclic amp. Annu. Rev. Biochem. 2003, 66, 807–822.
- Papavassiliou, A.G. The CREB/ATF family of transcription factors: Modulation by reversible phosphorylation. Anticancer Res. 1994, 14, 1801–1805.
- Niu, M.; Tabari, E.; Ni, P.; Su, Z. Towards a map of cis-regulatory sequences in the human genome. Nucleic Acids Res. 2018, 46, 5395–5409.
- D’Auria, F.; Centurione, L.; Centurione, M.A.; Angelini, A.; Di Pietro, R. Regulation of Cancer Cell Responsiveness to Ionizing Radiation Treatment by Cyclic AMP Response Element Binding Nuclear Transcription Factor. Front. Oncol. 2017, 7, 76.
- Lavrovsky, Y.; Schwartzman, M.L.; Levere, R.D.; Kappas, A.; Abraham, N.G. Identification of binding sites for transcription factors NF-kappa B and AP-2 in the promoter region of the human heme oxygenase 1 gene. Proc. Natl. Acad. Sci. USA 1994, 91, 5987–5991.
- Kogure, Y.; Kataoka, K. Genetic alterations in adult T-cell leukemia/lymphoma. Cancer Sci. 2017, 108, 1719–1725.
- Cataldi, A.; Rapino, M.; Centurione, L.; Sabatini, N.; Grifone, G.; Garaci, F.; Rana, R. NF-kB activation plays an antiapoptotic role in human leukemic K562 cells exposed to ionizing radiation. J. Cell. Biochem. 2003, 89, 956–963.
- Yu, H.; Aravindan, N.; Xu, J.; Natarajan, M. Inter- and intra-cellular mechanism of NF-kB-dependent survival advantage and clonal expansion of radio-resistant cancer cells. Cell. Signal. 2017, 31, 105–111.
- Mortezaee, K.; Najafi, M.; Farhood, B.; Ahmadi, A.; Shabeeb, D.; Musa, A.E. NF-κB targeting for overcoming tumor resistance and normal tissues toxicity. J. Cell. Physiol. 2019, 234, 17187–17204.
- Cataldi, A.; Di Giacomo, V.; Rapino, M.; Zara, S.; Rana, R.A. Ionizing Radiation Induces Apoptotic Signal Through Protein Kinase Cδ (delta) and Survival Signal Through Akt and Cyclic-Nucleotide Response Element-Binding Protein (CREB) in Jurkat T Cells. Biol. Bull. 2009, 217, 202–212.
- Katanyutanon, S.; Wu, R.; Wang, P. The effect of whole-body radiation on blood levels of gastrointestinal peptides in the rat. Int. J. Clin. Exp. Med. 2008, 1, 332–337.
- Chen, X.; Shen, B.; Xia, L.; Khaletzkiy, A.; Wong, J.Y.C.; Li, J.J.; Chu, D. Activation of nuclear factor κB in radioresistance of TP53-inactive human keratinocytes. Cancer Res. 2002, 62, 1213–1221.
- Chua, J.S.; Liew, H.P.; Guo, L.; Lane, D.P. Tumor-specific signaling to p53 is mimicked by Mdm2 inactivation in zebrafish: Insights from mdm2 and mdm4 mutant zebrafish. Oncogene 2015, 34, 5933–5941.
- Stewart-Ornstein, J.; Iwamoto, Y.; Miller, M.A.; Prytyskach, M.A.; Ferretti, S.; Holzer, P.; Kallen, J.; Furet, P.; Jambhekar, A.; Forrester, W.C.; et al. p53 dynamics vary between tissues and are linked with radiation sensitivity. Nat. Commun. 2021, 12, 898.
- Qi, J.-P.; Shao, S.-H.; Li, D.-D.; Zhou, G.-P. A dynamic model for the p53 stress response networks under ion radiation. Amino Acids 2007, 33, 75–83.
- Qi, J.; Shao, S.; Shen, Y. Cellular responding DNA damage: An improved modeling of P53 gene regulatory networks under ion radiation (IR). Appl. Math. Comput. 2008, 205, 73–83.
- Macaluso, M.; Montanari, M.; Cinti, C.; Giordano, A. Modulation of Cell Cycle Components by Epigenetic and Genetic Events. Semin. Oncol. 2005, 32, 452–457.
- Teufel, D.P.; Bycroft, M.; Fersht, A.R. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene 2009, 28, 2112–2118.
- Cataldi, A.; Di Giacomo, V.; Rapino, M.; Genovesi, D.; Rana, R.A. Cyclic Nucleotide Response Element Binding Protein (CREB) Activation Promotes Survival Signal in Human K562 Erythroleukemia Cells Exposed to Ionising Radiation/Etoposide Combined Treatment. J. Radiat. Res. 2006, 47, 113–120.
- Sokolov, M.; Neumann, R. Changes in gene expression as one of the key mechanisms involved in radiation-induced bystander effect (Review). Biomed. Rep. 2018, 9, 99–111.
- DeVries-Seimon, T.A.; Ohm, A.M.; Humphries, M.J.; Reyland, M.E. Induction of Apoptosis Is Driven by Nuclear Retention of Protein Kinase Cδ. J. Biol. Chem. 2007, 282, 22307–22314.
- Bluwstein, A.; Kumar, N.; Léger, K.; Traenkle, J.; van Oostrum, J.; Rehrauer, H.; Baudis, M.; Hottiger, M.O. PKC signaling prevents irradiation-induced apoptosis of primary human fibroblasts. Cell Death Dis. 2013, 4, e498.
- Martelli, A.M.; Evangelisti, C.; Nyakern, M.; Manzoli, F.A. Nuclear protein kinase C. Biochim. Biophys. Acta 2006, 1761, 542–551.
- Xue, Y.; Ren, J.; Gao, X.; Jin, C.; Wen, L.; Yao, X. GPS 2.0, a tool to predict kinase-specific phosphorylation sites in hierarchy. Mol. Cell. Proteomics 2008, 7, 1598–1606.
- Nickoloff, J.A.; Sharma, N.; Taylor, L. Clustered DNA Double-Strand Breaks: Biological Effects and Relevance to Cancer Radiotherapy. Genes 2020, 11, 99.
- Alam, M.; Kashyap, T.; Pramanik, K.K.; Singh, A.K.; Nagini, S.; Mishra, R. The elevated activation of NFκB and AP-1 is correlated with differential regulation of Bcl-2 and associated with oral squamous cell carcinoma progression and resistance. Clin. Oral Investig. 2017, 21, 2721–2731.
- Voboril, R.; Rychterova, V.; Voborilova, J.; Kubecova, M.; Fanta, J.; Dvorak, J. NF-κB/p65 expression before and after treatment in rectal cancer patients undergoing neoadjuvant (chemo)radiotherapy and surgery: Prognostic marker for disease progression and survival. Neoplasma 2016, 63, 462–470.
- Di Nisio, C.; Sancilio, S.; Di Giacomo, V.; Rapino, M.; Sancillo, L.; Genovesi, D.; Di Siena, A.; Rana, R.A.; Cataldi, A.; Di Pietro, R. Involvement of cyclic-nucleotide response element-binding family members in the radiation response of Ramos B lymphoma cells. Int. J. Oncol. 2016, 48, 28–36.
- Basu, S.; Rosenzweig, K.R.; Youmell, M.; Price, B.D. The DNA-Dependent Protein Kinase Participates in the Activation of NFκB Following DNA Damage. Biochem. Biophys. Res. Commun. 1998, 247, 79–83.
- Madhusoodhanan, R.; Natarajan, M.N.; Veeraraghavan, J.; Herman, T.S.; Aravindan, N. NFκB activity and transcriptional responses in human breast adenocarcinoma cells after single and fractionated irradiation. Cancer Biol. Ther. 2009, 8, 765–773.
- Ramos, P.M.M.; Pezuk, J.A.; Castro-Gamero, A.M.; Oliveira, H.F.; Scrideli, C.A.; Umezawa, K.; Tone, L.G. Antineoplastic Effects of NF-κB Inhibition by DHMEQ (Dehydroxymethylepoxyquinomicin) Alone and in Co-treatment with Radio-and Chemotherapy in Medulloblastoma Cell Lines. Anticancer. Agents Med. Chem. 2018, 18, 541–549.
- Samuel, T.; Fadlalla, K.; Gales, D.N.; Putcha, B.D.; Manne, U. Variable NF-κB pathway responses in colon cancer cells treated with chemotherapeutic drugs. BMC Cancer 2014, 14, 599.
- Chen, M.; Singh, A.K.; Repasky, E.A. Highlighting the Potential for Chronic Stress to Minimize Therapeutic Responses to Radiotherapy through Increased Immunosuppression and Radiation Resistance. Cancers 2020, 12, 3853.
- Nelson, E.C.; Cambio, A.J.; Yang, J.C.; Ok, J.-H.; Lara, P.N.; Evans, C.P. Clinical implications of neuroendocrine differentiation in prostate cancer. Prostate Cancer Prostatic 2007, 10, 6–14.
- Huang, Y.-H.; Zhang, Y.-Q.; Huang, J.-T. Neuroendocrine cells of prostate cancer: Biologic functions and molecular mechanisms. Asian J. Androl. 2019, 21, 291.
- Liu, M.; Guyot-Sionnest, P. Mechanism of silver(I)-assisted growth of gold nanorods and bipyramids. J. Phys. Chem. B 2005, 109, 22192–22200.
- Deng, X.; Liu, H.; Huang, J.; Cheng, L.; Keller, E.T.; Parsons, S.J.; Hu, C.-D. Ionizing Radiation Induces Prostate Cancer Neuroendocrine Differentiation through Interplay of CREB and ATF2: Implications for Disease Progression. Cancer Res. 2008, 68, 9663–9670.
- Di Pietro, R.; Fang, H.; Fields, K.; Miller, S.; Flora, M.; Petricoin, E.C.; Dveksler, G.; Rana, R.A.; Grimley, P.M. Peroxiredoxin Genes are Not Induced in Myeloid Leukemia Cells Exposed to Ionizing Radiation. Int. J. Immunopathol. Pharmacol. 2006, 19, 517–524.
- Rhee, S.G. Overview on Peroxiredoxin. Mol. Cells 2016, 39, 1–5.
- Wang, T.; Diaz, A.J.G.; Yen, Y. The role of peroxiredoxin II in chemoresistance of breast cancer cells. Breast Cancer Targ. Ther. 2014, 6, 73–80.
- Steven, A.; Seliger, B. Control of CREB expression in tumors: From molecular mechanisms and signal transduction pathways to therapeutic target. Oncotarget 2016, 7, 35454–35465.
- Jean, D.; Menashe, B.-E.; Bar-Eli, M. Regulation of tumor growth and metastasis of human melanoma by the CREB transcription factor family. Mol. Cell. Biochem. 2000, 212, 19–28.
- Alper, O.; Bergmann-Leitner, E.S.; Abrams, S.; Cho-Chung, Y.S. Apoptosis, growth arrest and suppression of invasiveness by CRE-decoy oligonucleotide in ovarian cancer cells: Protein kinase A downregulation and cytoplasmic export of CRE-binding proteins. Mol. Cell. Biochem. 2001, 218, 55–63.
- Steven, A.; Leisz, S.; Massa, C.; Iezzi, M.; Lattanzio, R.; Lamolinara, A.; Bukur, J.; Müller, A.; Hiebl, B.; Holzhausen, H.-J.; et al. HER-2/neu Mediates Oncogenic Transformation via Altered CREB Expression and Function. Mol. Cancer Res. 2013, 11, 1462–1477.
- Xie, F.; Li, B.X.; Kassenbrock, A.; Xue, C.; Wang, X.; Qian, D.Z.; Sears, R.C.; Xiao, X. Identification of a Potent Inhibitor of CREB-Mediated Gene Transcription with Efficacious in Vivo Anticancer Activity. J. Med. Chem. 2015, 58, 5075–5087.
- Tsolou, A.; Liousia, M.; Kalamida, D.; Pouliliou, S.; Giatromanolaki, A.; Koukourakis, M. Inhibition of IKK-NFκB pathway sensitizes lung cancer cell lines to radiation. Cancer Biol. Med. 2017, 14, 293–301.
- Molavi Pordanjani, S.; Jalal Hosseinimehr, S. The Role of NF-κB Inhibitors in Cell Response to Radiation. Curr. Med. Chem. 2016, 23, 3951–3963.
- Hara, K.; Takeda, A.; Tsurugai, Y.; Saigusa, Y.; Sanuki, N.; Eriguchi, T.; Maeda, S.; Tanaka, K.; Numata, K. Radiotherapy for Hepatocellular Carcinoma Results in Comparable Survival to Radiofrequency Ablation: A Propensity Score Analysis. Hepatology 2019, 69, 2533–2545.
- Fuchs-Tarlovsky, V. Role of antioxidants in cancer therapy. Nutrition 2013, 29, 15–21.
- Impicciatore, G.; Sancilio, S.; Miscia, S.; Di Pietro, R. Nutlins and Ionizing Radiation in Cancer Therapy. Curr. Pharm. Des. 2010, 16, 1427–1442.
- Nayak, S.K.; Khatik, G.L.; Narang, R.; Monga, V.; Chopra, H.K. p53-Mdm2 Interaction Inhibitors as Novel Nongenotoxic Anticancer Agents. Curr. Cancer Drug Targ. 2017, 18, 749–772.
- Zauli, G.; Celeghini, C.; Melloni, E.; Voltan, R.; Ongari, M.; Tiribelli, M.; di Iasio, M.G.; Lanza, F.; Secchiero, P. The sorafenib plus nutlin-3 combination promotes synergistic cytotoxicity in acute myeloid leukemic cells irrespectively of FLT3 and p53 status. Haematologica 2012, 97, 1722–1730.
- Gu, L.; Zhu, N.; Findley, H.W.; Zhou, M. MDM2 antagonist nutlin-3 is a potent inducer of apoptosis in pediatric acute lymphoblastic leukemia cells with wild-type p53 and overexpression of MDM2. Leukemia 2008, 22, 730–739.
- Zauli, G.; Voltan, R.; Bosco, R.; Melloni, E.; Marmiroli, S.; Rigolin, G.M.; Cuneo, A.; Secchiero, P. Dasatinib Plus Nutlin-3 Shows Synergistic Antileukemic Activity in Both p53 wild-type and p53 mutated B Chronic Lymphocytic Leukemias by Inhibiting the Akt Pathway. Clin. Cancer Res. 2011, 17, 762–770.
- Roh, J.L.; Ko, J.H.; Moon, S.J.; Ryu, C.H.; Choi, J.Y.; Koch, W.M. The p53-reactivating small-molecule RITA enhances cisplatin-induced cytotoxicity and apoptosis in head and neck cancer. Cancer Lett. 2012, 325, 35–41.
- Wiman, K.G. Pharmacological reactivation of mutant p53: From protein structure to the cancer patient. Oncogene 2010, 29, 4245–4252.
- Hientz, K.; Mohr, A.; Bhakta-Guha, D.; Efferth, T. The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 2017, 8, 8921–8946.