Neutrophils and platelets exhibit a diverse repertoire of functions in thromboinflammatory conditions such as stroke. Neutrophils can enable, as well as resolve, cerebrovascular inflammation via many effector functions including neutrophil extracellular traps, serine proteases and reactive oxygen species, and pro-resolving endogenous molecules such as Annexin A1. Like neutrophils, platelets also engage in pro- as well as anti-inflammatory roles in regulating cerebrovascular inflammation. These anucleated cells are at the core of stroke pathogenesis and can trigger an ischemic event via adherence to the hypoxic cerebral endothelial cells culminating in aggregation and clot formation.
- neutrophils
- platelets
- stroke
- annexin A1
- resolution
- thromboinflammation
1. Introduction
Neutrophils and platelets are key players in ischemic brain injury and its resolution [1][2][3][4]. Resolution is the physiological ability of the body to achieve homeostasis after infection or inflammation. However, in chronic inflammation, where there is an excessive and persistent inflammatory response, the process of resolution is hampered [5][6]. Acute cerebral ischemia induces a strong immune response resulting in recruitment of several subsets of leukocytes (mainly neutrophils), activation of platelets, and coagulation cascade and upregulation of cell adhesion molecules and cytokines [7]. Neutrophils and platelets are known for their ability to produce proinflammatory/prothrombotic mediators, thereby forming an important link between inflammation and thrombosis, a phenomenon referred to as “thromboinflammation” [1][8][9].
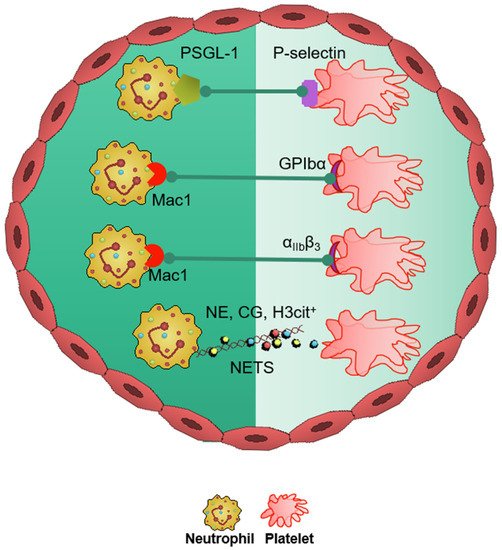
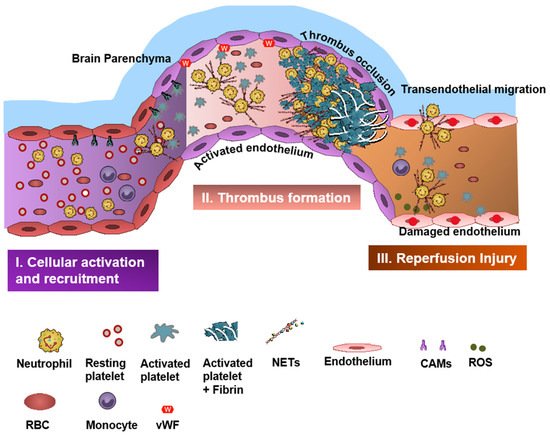
2. Neutrophils in Stroke
3. Neutrophil Serine Proteases and Thromboinflammation
4. Neutrophil-Dependent Oxidative Stress and IS
5. Platelets in Stroke
6. Therapeutics in Thromboinflammation
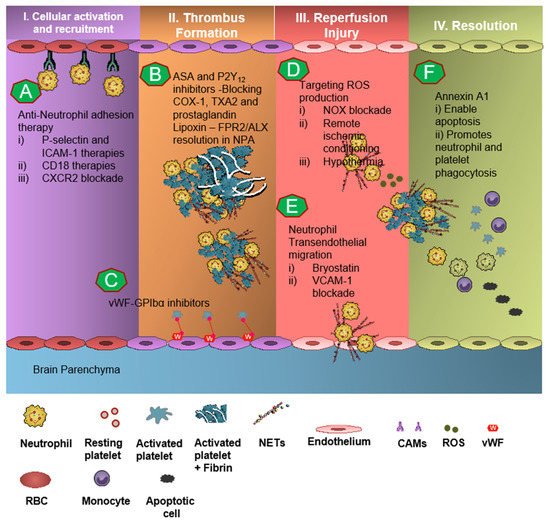
7. Targeting Neutrophil-Dependent Thromboinflammation
Neutrophil recruitment to the ischemic site and adhesion to brain endothelial cells is enabled by P-selectin and ICAM-1 [51][52][53]. The anti-neutrophil adhesion strategy targeting P-selectin and ICAM-1 was proven to diminish neutrophil recruitment and transmigration at the site of cerebral I/R, thereby resulting in attenuation of thromboinflammation [52][53]. CD18 (leukocyte counter-ligand to endothelial intracellular adhesion molecule-1) knockout mice conferred cerebrovascular protection in a murine model of IS, but not to CD18-deficient animals with permanent middle cerebral artery occlusion, suggesting anti-neutrophil adhesion strategies should be further tested for the management of stroke [52]. However, Enlimomab, a murine ICAM-1 antibody that is known to reduce leukocyte adhesion and infarct size in experimental stroke studies, was not effective in earlier clinical trials, with more adverse events such as infections and fever compared to the placebo [54]. Studies targeting anti-E-selectin, anti-L-selectin, and chemokine receptors had no response to minimal response in animal models of experimental IS [55].
8. Targeting Platelet-Dependent Thromboinflammation
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines9121945
References
- Ansari, J.; Senchenkova, E.Y.; Vital, S.A.; Al Yafeai, Z.; Kaur, G.; Sparkenbaugh, E.M.; Orr, A.; Pawlinski, R.; Hebbel, R.P.; Granger, D.N.; et al. Targeting AnxA1/Fpr2/ALX Pathway Regulates Neutrophil Function Promoting Thrombo-Inflammation Resolution in Sickle Cell Disease. Blood 2021, 137, 1538–1549.
- Ansari, J.; Gavins, F.N.E. The impact of thrombo-inflammation on the cerebral microcirculation. Microcirculation 2021, 28, e12689.
- Senchenkova, E.Y.; Ansari, J.; Becker, F.; Vital, S.A.; Al-Yafeai, Z.; Sparkenbaugh, E.M.; Pawlinski, R.; Stokes, K.Y.; Carroll, J.L.; Dragoi, A.M.; et al. A Novel Role for the AnxA1-Fpr2/ALX Signaling Axis as a Key Regulator of Platelet Function to Promote Resolution of Inflammation. Circulation 2019, 140, 319–335.
- Vital, S.A.; Becker, F.; Holloway, P.M.; Russell, J.; Perretti, M.; Granger, D.N.; Gavins, F.N. Formyl-Peptide Receptor 2/3/Lipoxin A4 Receptor Regulates Neutrophil-Platelet Aggregation and Attenuates Cerebral Inflammation: Impact for Therapy in Cardiovascular Disease. Circulation 2016, 133, 2169–2179.
- Perretti, M.; Leroy, X.; Bland, E.J.; Montero-Melendez, T. Resolution Pharmacology: Opportunities for Therapeutic Innovation in Inflammation. Trends Pharmacol. Sci. 2015, 36, 737–755.
- Serhan, C.N. Pro-resolving lipid mediators are leads for resolution physiology. Nature 2014, 510, 92–101.
- Stoll, G.; Kleinschnitz, C.; Nieswandt, B. Combating innate inflammation: A new paradigm for acute treatment of stroke? Ann. N. Y. Acad. Sci. 2010, 1207, 149–154.
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896.
- De Meyer, S.F.; Denorme, F.; Langhauser, F.; Geuss, E.; Fluri, F.; Kleinschnitz, C. Thromboinflammation in Stroke Brain Damage. Stroke 2016, 47, 1165–1172.
- Cerletti, C.; Tamburrelli, C.; Izzi, B.; Gianfagna, F.; de Gaetano, G. Platelet-leukocyte interactions in thrombosis. Thromb. Res. 2012, 129, 263–266.
- Arumugam, T.V.; Salter, J.W.; Chidlow, J.H.; Ballantyne, C.M.; Kevil, C.G.; Granger, D.N. Contributions of LFA-1 and Mac-1 to brain injury and microvascular dysfunction induced by transient middle cerebral artery occlusion. Am. J. Physiol. Heart Circ. Physiol. 2004, 287, H2555–H2560.
- Li, N.; Mao, D.; Lü, S.; Tong, C.; Zhang, Y.; Long, M. Distinct binding affinities of Mac-1 and LFA-1 in neutrophil activation. J. Immunol. 2013, 190, 4371–4381.
- Goldsmith, H.L.; Spain, S. Margination of leukocytes in blood flow through small tubes. Microvasc. Res. 1984, 27, 204–222.
- Enzmann, G.; Kargaran, S.; Engelhardt, B. Ischemia-reperfusion injury in stroke: Impact of the brain barriers and brain immune privilege on neutrophil function. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418794184.
- Ansari, J.; Gavins, F.N.E. Ischemia-Reperfusion Injury in Sickle Cell Disease: From Basics to Therapeutics. Am. J. Pathol. 2019, 189, 706–718.
- Ritter, L.S.; Stempel, K.M.; Coull, B.M.; McDonagh, P.F. Leukocyte-platelet aggregates in rat peripheral blood after ischemic stroke and reperfusion. Biol. Res. Nurs. 2005, 6, 281–288.
- Harris, M.G.; Hulseberg, P.; Ling, C.; Karman, J.; Clarkson, B.D.; Harding, J.S.; Zhang, M.; Sandor, A.; Christensen, K.; Nagy, A.; et al. Immune privilege of the CNS is not the consequence of limited antigen sampling. Sci. Rep. 2014, 4, 4422.
- Ousman, S.S.; Kubes, P. Immune surveillance in the central nervous system. Nat. Neurosci. 2012, 15, 1096–1101.
- Manda-Handzlik, A.; Demkow, U. The Brain Entangled: The Contribution of Neutrophil Extracellular Traps to the Diseases of the Central Nervous System. Cells 2019, 8, 1477.
- Allen, C.; Thornton, P.; Denes, A.; McColl, B.W.; Pierozynski, A.; Monestier, M.; Pinteaux, E.; Rothwell, N.J.; Allan, S.M. Neutrophil cerebrovascular transmigration triggers rapid neurotoxicity through release of proteases associated with decondensed DNA. J. Immunol. 2012, 189, 381–392.
- Buck, B.H.; Liebeskind, D.S.; Saver, J.L.; Bang, O.Y.; Yun, S.W.; Starkman, S.; Ali, L.K.; Kim, D.; Villablanca, J.P.; Salamon, N.; et al. Early neutrophilia is associated with volume of ischemic tissue in acute stroke. Stroke 2008, 39, 355–360.
- Quan, K.; Wang, A.; Zhang, X.; Meng, X.; Chen, P.; Li, H.; Wang, Y. Neutrophil to lymphocyte ratio and adverse clinical outcomes in patients with ischemic stroke. Ann. Transl. Med. 2021, 9, 1047.
- Ying, Y.; Yu, F.; Luo, Y.; Feng, X.; Liao, D.; Wei, M.; Li, X.; Huang, Q.; Liu, Z.; Zhang, L.; et al. Neutrophil-to-Lymphocyte Ratio as a Predictive Biomarker for Stroke Severity and Short-Term Prognosis in Acute Ischemic Stroke With Intracranial Atherosclerotic Stenosis. Front. Neurol. 2021, 12, 705949.
- Jiang, X.; Andjelkovic, A.V.; Zhu, L.; Yang, T.; Bennett, M.V.L.; Chen, J.; Keep, R.F.; Shi, Y. Blood-brain barrier dysfunction and recovery after ischemic stroke. Prog. Neurobiol. 2018, 163–164, 144–171.
- Rosell, A.; Cuadrado, E.; Ortega-Aznar, A.; Hernandez-Guillamon, M.; Lo, E.H.; Montaner, J. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 2008, 39, 1121–1126.
- Faraday, N.; Schunke, K.; Saleem, S.; Fu, J.; Wang, B.; Zhang, J.; Morrell, C.; Dore, S. Cathepsin G-dependent modulation of platelet thrombus formation in vivo by blood neutrophils. PLoS ONE 2013, 8, e71447.
- Woloszynek, J.C.; Hu, Y.; Pham, C.T. Cathepsin G-regulated release of formyl peptide receptor agonists modulate neutrophil effector functions. J. Biol. Chem. 2012, 287, 34101–34109.
- Papayannopoulos, V.; Staab, D.; Zychlinsky, A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving Dnase therapy. PloS ONE 2011, 6, e28526.
- Pham, C.T. Neutrophil serine proteases: Specific regulators of inflammation. Nat. Rev. Immunol. 2006, 6, 541–550.
- Burgener, S.S.; Leborgne, N.G.F.; Snipas, S.J.; Salvesen, G.S.; Bird, P.I.; Benarafa, C. Cathepsin G Inhibition by Serpinb1 and Serpinb6 Prevents Programmed Necrosis in Neutrophils and Monocytes and Reduces GSDMD-Driven Inflammation. Cell Rep. 2019, 27, 3646–3656.
- Sun, R.; Iribarren, P.; Zhang, N.; Zhou, Y.; Gong, W.; Cho, E.H.; Lockett, S.; Chertov, O.; Bednar, F.; Rogers, T.J.; et al. Identification of neutrophil granule protein cathepsin G as a novel chemotactic agonist for the G protein-coupled formyl peptide receptor. J. Immunol. 2004, 173, 428–436.
- Chan, P.H. Oxygen radicals in focal cerebral ischemia. Brain Pathol. 1994, 4, 59–65.
- Lorenzano, S.; Rost, N.S.; Khan, M.; Li, H.; Batista, L.M.; Chutinet, A.; Green, R.E.; Thankachan, T.K.; Thornell, B.; Muzikansky, A.; et al. Early molecular oxidative stress biomarkers of ischemic penumbra in acute stroke. Neurology 2019, 93, e1288–e1298.
- Cook-Mills, J.M.; Marchese, M.E.; Abdala-Valencia, H. Vascular cell adhesion molecule-1 expression and signaling during disease: Regulation by reactive oxygen species and antioxidants. Antioxid. Redox Signal. 2011, 15, 1607–1638.
- Rodrigo, R.; Fernandez-Gajardo, R.; Gutierrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714.
- Hoda, M.N.; Siddiqui, S.; Herberg, S.; Periyasamy-Thandavan, S.; Bhatia, K.; Hafez, S.S.; Johnson, M.H.; Hill, W.D.; Ergul, A.; Fagan, S.C.; et al. Remote ischemic perconditioning is effective alone and in combination with intravenous tissue-type plasminogen activator in murine model of embolic stroke. Stroke 2012, 43, 2794–2799.
- Han, Z.; Liu, X.; Luo, Y.; Ji, X. Therapeutic hypothermia for stroke: Where to go? Exp. Neurol. 2015, 272, 67–77.
- Ohkura, N.; Hiraishi, S.; Itabe, H.; Hamuro, T.; Kamikubo, Y.; Takano, T.; Matsuda, J.; Horie, S. Oxidized phospholipids in oxidized low-density lipoprotein reduce the activity of tissue factor pathway inhibitor through association with its carboxy-terminal region. Antioxid. Redox Signal. 2004, 6, 705–712.
- Rawish, E.; Nording, H.; Münte, T.; Langer, H.F. Platelets as Mediators of Neuroinflammation and Thrombosis. Front. Immunol. 2020, 11, 548631.
- Kehrel, B.E.; Fender, A.C. Resolving Thromboinflammation in the Brain After Ischemic Stroke? Circulation 2016, 133, 2128–2131.
- Lievens, D.; Zernecke, A.; Seijkens, T.; Soehnlein, O.; Beckers, L.; Munnix, I.C.; Wijnands, E.; Goossens, P.; van Kruchten, R.; Thevissen, L.; et al. Platelet CD40L mediates thrombotic and inflammatory processes in atherosclerosis. Blood 2010, 116, 4317–4327.
- Perdomo, J.; Leung, H.H.L.; Ahmadi, Z.; Yan, F.; Chong, J.J.H.; Passam, F.H.; Chong, B.H. Neutrophil activation and NETosis are the major drivers of thrombosis in heparin-induced thrombocytopenia. Nat. Commun. 2019, 10, 1322.
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654.
- Clark, S.R.; Ma, A.C.; Tavener, S.A.; McDonald, B.; Goodarzi, Z.; Kelly, M.M.; Patel, K.D.; Chakrabarti, S.; McAvoy, E.; Sinclair, G.D.; et al. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat. Med. 2007, 13, 463–469.
- Rana, A.; Westein, E.; Niego, B.; Hagemeyer, C.E. Shear-Dependent Platelet Aggregation: Mechanisms and Therapeutic Opportunities. Front. Cardiovasc. Med. 2019, 6, 141.
- Meyer, S.F.D.; Stoll, G.; Wagner, D.D.; Kleinschnitz, C. von Willebrand Factor. Stroke 2012, 43, 599–606.
- Shankaran, H.; Alexandridis, P.; Neelamegham, S. Aspects of hydrodynamic shear regulating shear-induced platelet activation and self-association of von Willebrand factor in suspension. Blood 2003, 101, 2637–2645.
- Li, Y.; Choi, H.; Zhou, Z.; Nolasco, L.; Pownall, H.J.; Voorberg, J.; Moake, J.L.; Dong, J.F. Covalent regulation of ULVWF string formation and elongation on endothelial cells under flow conditions. J. Thromb. Haemost. 2008, 6, 1135–1143.
- Ansari, J.; Kaur, G.; Gavins, F.N.E. Therapeutic Potential of Annexin A1 in Ischemia Reperfusion Injury. Int. J. Mol. Sci. 2018, 19, 1211.
- Vital, S.A.; Senchenkova, E.Y.; Ansari, J.; Gavins, F.N.E. Targeting AnxA1/Formyl Peptide Receptor 2 Pathway Affords Protection against Pathological Thrombo-Inflammation. Cells 2020, 9, 2473.
- Drieu, A.; Levard, D.; Vivien, D.; Rubio, M. Anti-inflammatory treatments for stroke: From bench to bedside. Ther. Adv. Neurol. Disord. 2018, 11, 1756286418789854.
- Prestigiacomo, C.J.; Kim, S.C.; Connolly, E.S., Jr.; Liao, H.; Yan, S.F.; Pinsky, D.J. CD18-mediated neutrophil recruitment contributes to the pathogenesis of reperfused but not nonreperfused stroke. Stroke 1999, 30, 1110–1117.
- Suzuki, H.; Hayashi, T.; Tojo, S.J.; Kitagawa, H.; Kimura, K.; Mizugaki, M.; Itoyama, Y.; Abe, K. Anti-P-selectin antibody attenuates rat brain ischemic injury. Neurosci. Lett. 1999, 265, 163–166.
- Enlimomab Acute Stroke Trial, I. Use of anti-ICAM-1 therapy in ischemic stroke: Results of the Enlimomab Acute Stroke Trial. Neurology 2001, 57, 1428–1434.
- Jickling, G.C.; Liu, D.; Ander, B.P.; Stamova, B.; Zhan, X.; Sharp, F.R. Targeting neutrophils in ischemic stroke: Translational insights from experimental studies. J. Cereb. Blood Flow Metab. 2015, 35, 888–901.
- Hankey, G.J. The benefits of aspirin in early secondary stroke prevention. Lancet 2016, 388, 312–314.
- Awtry, E.H.; Loscalzo, J. Aspirin. Circulation 2000, 101, 1206–1218.
- Rothwell, P.M.; Giles, M.F.; Chandratheva, A.; Marquardt, L.; Geraghty, O.; Redgrave, J.N.; Lovelock, C.E.; Binney, L.E.; Bull, L.M.; Cuthbertson, F.C.; et al. Effect of urgent treatment of transient ischaemic attack and minor stroke on early recurrent stroke (EXPRESS study): A prospective population-based sequential comparison. Lancet 2007, 370, 1432–1442.
- Meadows, T.A.; Bhatt, D.L. Clinical aspects of platelet inhibitors and thrombus formation. Circ. Res. 2007, 100, 1261–1275.
- Hackam, D.G.; Spence, J.D. Antiplatelet Therapy in Ischemic Stroke and Transient Ischemic Attack. Stroke 2019, 50, 773–778.