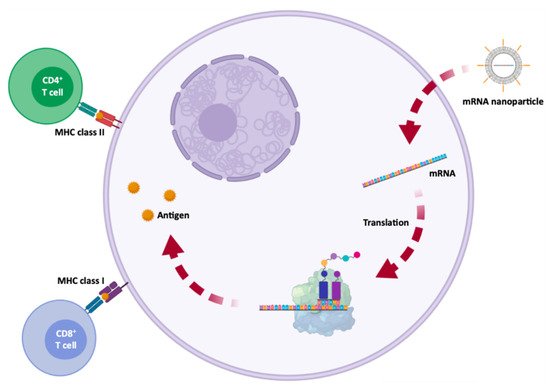
Currently, mRNA appears as a very promising and innovative therapeutic approach for diseases associated with functional loss of proteins, through the administration of a synthetic mRNA, which promotes the reestablishment of protein levels and restores its function. Moreover, mRNA can create new cellular functions, for example for passive immunization, allowing to stimulate the immune system, through the translation of antigenic mRNA for specific cell recognition (e.g., cancer cells) or antibody production. The fact that a relatively small amount of encoded antigen, from a synthetic mRNA, can be sufficient to obtain robust signs of efficacy, is one of the main advantages of using this biomolecule in immunotherapy. However, the global success of such mRNA-based treatments depends on a high number of these biomolecules and an effective in vivo delivery to target cells involved in a given disease. After proving that in vivo mRNA administration is possible and viable, the concept of using mRNA as a therapeutic basis was readily accepted and used in a variety of diseases, including diabetes, HIV infection, anemia, hemophilia, myocardial infarction, cancer, asthma, metabolic disorders, fibrosis, skeletal degeneration and neurological disorders, such as Friedreich’s ataxia and Alzheimer’s disease.
- mRNA
- protein replacement therapies
- immunotherapy
- gene editing
1. Protein Replacement Therapies
| Therapeutic Approach | Objective/Function |
|---|---|
| Protein Replacement | Restore function, increase expression or replace protein in rare monogenic diseases |
| Cell reprogramming | Modulate cellular behavior by expressing transcription and/or growth factors |
| Immunotherapies | Elicit specific immune responses against target cells, for example through therapeutic antibodies |
2. Immunotherapy
| mRNA Vaccines | Structure | Advantages | Disadvantages | References |
|---|---|---|---|---|
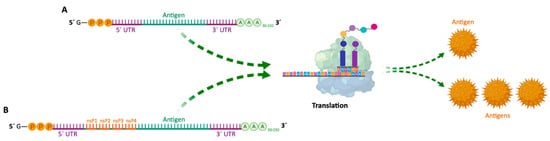
| Non-amplifying mRNA Vaccines | Basic structure of the mRNA, with a coding region for the desired antigens. | - Relatively small mRNA size (~2–3 kb). - Absence of additional proteins, minimizing unwanted immune interactions. - Relatively easy to produce and amplify. - Simplified sequence engineering. - Direct antigen expression. |
- Potential toxicity from modified nucleotides. - Short duration of expression. - Need for high RNA doses. - Low antigen quantity. |
[2][15] |
| SAM Vaccines | Encode a manipulated RNA virus genome (replicon). It generally contains two different protein coding regions, one encoding nonstructural proteins involved in mRNA capping and replication, and the other in antigen expression. | - High yield of target antigen. - Enhanced and prolonged antigen expression. - Lower effective RNA doses (more safe). - Intrinsic adjuvant effect. - Potential apoptosis of vaccine-carrying cells due to vaccine self-amplification (enhanced cross-presentation). - Option for single-vector delivery of multiple or complex antigens. |
- RNA replicons are not able to tolerate many of the synthetic nucleotide modifications and sequence alterations. - Inclusion of unrelated proteins, which may increase unwanted immunogenicity. - Large replicon size (~10 kb), decreasing cell internalization efficiency. - Interaction between nsPs and host factors yet to be addressed. - Longer RNA length (more difficult production). - Potential elevated inflammation. |
[2][21][4][15][24] |
| Name | Therapetic Modality | Protein Target | Administration Method | Delivery Vehicle | Disease | Sponsor Institution | ClinicalTrials.gov Identifer | Phase | Therapeutic Approach | References |
|---|---|---|---|---|---|---|---|---|---|---|
| MRT5005 | mRNA | CFTR | Inhalation | LNPs | Cystic fibrosis | Translate Bio | NCT03375047 | I/II | Protein Replacement | [25] |
| AZD8601 | mRNA | VEGF-A | Intracardiac injection | Naked mRNA | Heart failure | AstraZeneca | NCT03370887 | II | Cell reprogramming | [26] |
| CV7201 | mRNA | Rabies virus glycoprotein | I.D or I.M | RNActive, protamine | Rabies | CureVac | NCT02241135 | I | Immunotherapy | [27] |
| CV7202 | mRNA | Rabies virus glycoprotein | I.M | LNPs | Rabies | NCT03713086 | I | Immunotherapy | [21] | |
| CV9201 | mRNA | TAAs | I.D | RNActive, protamine | NSCLC | NCT00923312 | I/II | Immunotherapy | [28] | |
| CV9202 | mRNA | TAAs | I.D | RNActive, protamine | NSCLC | NCT03164772 | I/II | Immunotherapy | [2] | |
| CV9104 | mRNA | TAAs | I.D | RNActive, protamine | Prostate carcinoma | NCT02140138 | II | Immunotherapy | ||
| HARE-40 | mRNA | HPV antigen CD40 | I.D | Naked RNA | HPV-driven squamous cell carcinoma |
BioNTech | NCT03418480 | I/II | Immunotherapy | |
| Lipo-MERIT | mRNA | TAAs: NYESO-1, MAGE-A3, tyrosinase, and TPTE | I.V | Lipo-MERIT, DOTMA(DOTAP)/ DOPE lipoplex |
Advanced melanoma | NCT02410733 | I | Immunotherapy | ||
| IVAC | mRNA | 3 TAAs selected from a warehouse and p53 RNA; Neo-Ag based on NGS screening | I.V | Lipo-MERIT, DOTMA(DOTAP)/DOPE lipoplex |
TNBC | BioNTech | NCT02316457 | I | Immunotherapy | [2] |
| RBL001/RBL002 | mRNA | TAAs | Ultrasound guided I.N |
Naked mRNA | Melanoma | NCT01684241 | I | Immunotherapy | ||
| IVAC MUTANOME | mRNA | Neo-Ag | Ultrasound guided I.N |
Naked mRNA | Melanoma | NCT02035956 | I | Immunotherapy | ||
| RO7198457 | mRNA | Neo-Ag | I.V | Naked mRNA | Melanoma; NSCLC; Bladder cancer | NCT03289962 | I | Immunotherapy | ||
| mRNA-1325 | mRNA | Zika virus antigen | I.D | LNPs | Zika virus | Moderna | NCT03014089 | I | Immunotherapy | |
| mRNA-1653 | mRNA | hMPV and hPIV type 3 vaccine | I.D | LNPs | hMPV and hPIV infection |
NCT03392389 | I | Immunotherapy | ||
| VAL-506440 | mRNA | H10N8 antigen | I.D | LNPs | Influenza | Moderna | NCT03076385 | I | Immunotherapy | [29] |
| VAL-339851 | mRNA | H7 influenza antigen | I.D | LNPs | Influenza | NCT03345043 | I | Immunotherapy | ||
| mRNA-1647/1443 | mRNA | CMV glycoprotein H pentamer complex | I.D | LNPs | CMV infection | NCT03382405 | I | Immunotherapy | [2] | |
| mRNA-2416 | mRNA | Human OX40L | I.D | LNPs | Solid tumor malignancies or lymphoma |
NCT03323398 | I | Immunotherapy | ||
| mRNA-4157 | mRNA | Neo-Ag | Intratumoral | LNPs | Solid tumor | NCT03313778 | I | Immunotherapy | ||
| mRNA-4650 | mRNA | Neo-Ag | I.M | Naked mRNA | Melanoma; Colon cancer; GI cancer; Genitourinary cancer; HCC |
NCT03480152 | I/II | Immunotherapy | [30][31] | |
| mRNA-1388 | mRNA | VAL-181388 | I.M | LNPs | CHIKV | NCT03325075 | I | Immunotherapy | [32] | |
| mRNA-2752 | mRNA | OX40L, IL-23, and IL-36γ | Intratumoral | LNPs | Solid tumor or lymphoma | Moderna/AstraZeneca | NCT03739931 | I | Immunotherapy | [21] |
| iHIVARNA-01 | mRNA | Trimix (CD40L, CD70 and caTLR4 RNA—mRNA-transfected) | I.N | Naked mRNA | HIV infection | Hospital Clínic de Barcelona |
NCT02413645 | I | Immunotherapy | [33] |
| mRNA | I.N | Naked mRNA | HIV infection | Erasmus Medical Center | NCT02888756 | II | Immunotherapy | [34] | ||
| - | mRNA | CT7, MAGE-A3, and WT1 mRNA-electroporated LCs | I.D | DC-loaded mRNA | Malignant melanoma | Memorial Sloan Kettering Cancer Center |
NCT01995708 | I | Immunotherapy | [2] |
| - | mRNA | HIV-1 Gag- and Nef-transfected DCs | I.D | DC-loaded mRNA | HIV infection | Massachusetts General Hospital |
NCT00833781 | I/II | Immunotherapy | [35] |
| - | mRNA | Neo-Ag | S.C | Naked mRNA | Solid tumor malignancies or lymphoma |
Changhai Hospital Stemirna Therapeutics |
NCT03468244 | N.A | Immunotherapy | [2] |
| - | mRNA | TAA for melanoma (Melan-A, MAGE-A1, MAGE-A3, survivin, GP100, and tyrosinase) |
I.D | Naked mRNA | Melanoma | University Hospital Tuebingen |
NCT00204516 | I/II | Immunotherapy | [32] |
| - | mRNA | TAA-transfected DC | I.D or I.N | DC-loaded mRNA | Malignant melanoma | Oslo University Hospital |
NCT01278940 | I/II | Immunotherapy | [36] |
| - | mRNA | I.D | DC-loaded mRNA | Prostate cancer | NCT01278914 | I/II | Immunotherapy | [2] | ||
| AVX601 | Replicon | Alphavirus replicon vaccine expressing CMV genes |
I.M or S.C | - | CMV | AlphaVax | NCT00439803 | I | Immunotherapy | [2] |
| AVX502 | Replicon | Alphavirus replicon vaccine expressing an influenza HA protein |
I.M or S.C | - | Influenza | NCT00440362; NCT00706732 |
I/II | Immunotherapy | ||
| AVX101 | Replicon | Alphavirus replicon, HIV-1 subtype C Gag vaccine | I.M or S.C | - | HIV infections | NCT00097838; NCT00063778 | I | Immunotherapy | [37] | |
| AVX701 | Replicon | Alphavirus replicon encoding the protein | I.M or S.C | - | Colon cancer; CRC; Breast cancer; Lung cancer; Pancreatic cancer |
NCT01890213; NCT00529984 |
I/II | Immunotherapy | [2] | |
| NY-ESO-1 | CRISPR-Cas9 | PD-1 and TCR | Ex vivo | Autologous T cells | Multiple myeloma; Synovial sarcoma; Melanoma |
University of Pennsylvania | NCT03399448 | I | Gene Editing | [2] |
| CRISPR/TALEN-HPV E6/E7 | CRISPR/Cas9, TALEN | E6 and E7 | N.A | Plasmid DNA in gel | Cervical intraepithelial neoplasia | First Affiliated Hospital, Sun Yat-Sen University | NCT03057912 | I | Gene Editing | [38][39] |
| CTX001 | CRISPR-Cas9 | BCL11A | Ex vivo | Modified CD34+ hHSPCs | ß-thalassemia | Vertex Pharmaceuticals Incorporated | NCT03655678 | I/II | Gene Editing | [2] |
| - | CRISPR-Cas9 | PD-1 and TCR | Ex vivo | CAR-T cells | Mesothelin positive multiple solid tumors | Chinese PLA General Hospital | NCT03545815 | I | Gene Editing | |
| - | CRISPR-Cas9 | CD19 and CD20 | Ex vivo | Dual specificity CAR-T cells | ß cell leukemia and lymphoma | NCT03398967 | I/II | Gene Editing | ||
| UCART019 | CRISPR-Cas9 | CD19 | Ex vivo | CAR-T cells | ß cell leukemia and lymphoma | NCT03166878 | I/II | Gene Editing | [40] | |
| - | CRISPR-Cas9 | PD-1 | Ex vivo | Cytotoxic T lymphocytes | EBV-associated malignancies |
Yang Yang | NCT03044743 | I/II | Gene Editing | [41][42] |
| SB-728mR-HSPC | ZFN mRNA | CCR5 | Ex vivo (mRNA) | CD34+ hHSPCs | HIV | City of Hope Medical Center | NCT02500849 | I | Gene Editing | [43] |
| SB-728mR-T | ZFN mRNA | CCR5 | Ex vivo (mRNA) | T cells | HIV | Sangamo Therapeutics | NCT02225665 | I/II | Gene Editing | [44] |
| Lipid Name | Role | Abbreviation | Molar Lipid Ratios (%) (Ionizable Cationic Lipid:Neutral Lipid:Cholesterol:PEG-ylated Lipid) |
|---|---|---|---|
| BNT162b2 vaccine | |||
| 4-hydroxybutyl)azanediyl)bis(hexane-6,1-diyl)bis(2-hexyldecanoate | ionizable cationic lipid | ALC-0315 | 46.3:9.4:42.7:1.6 |
| 1,2-Distearoyl-sn-glycero-3-phosphocholine | helper lipid | DSPC | |
| cholesterol | helper lipid | Chol | |
| 2-[(polyethylene glycol)-2000]-N,N-ditetradecylacetamide | PEG-lipid | ALC-0159 | |
| mRNA-1273 vaccine | |||
| heptadecan-9-yl 8-((2-hydroxyethyl)[6-oxo-6-(undecyloxy)hexyl]amino)octanoate | ionizable cationic lipid | SM-102 | 50:10:38.5:1.5 |
| 1,2-distearoyl-sn-glycero-3-phosphocholine | helper lipid | DSPC | |
| cholesterol | helper lipid | Chol | |
| 1,2-Dimyristoyl-rac-glycero-3-methoxypolyethylene glycol-2000 | PEG-lipid | PEG2000-DMG | |
| Name | Therapetic Modality | Protein Target | Administration Method | Delivery Vehicle | Developer | ClinicalTrials.gov Identifer, EU Clinical Trials Register or Chinese Clinical Trial Register | Phase |
|---|---|---|---|---|---|---|---|
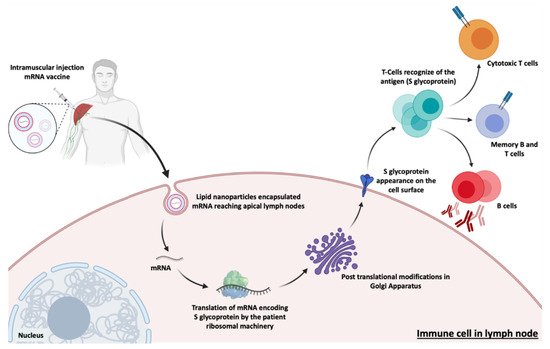
| mRNA-1273 | mRNA | Spike Glycoprotein | Intramuscular | LNP | Moderna/NIAID | EUCTR2021-002327-38-NL | IV |
| BNT162b2 | mRNA | RBD/Spike Glycoprotein | Intramuscular | LNP | Pfizer/BioNTech + Fosun Pharma | NCT04760132 | IV |
| CVnCoV Vaccine | mRNA | Spike Glycoprotein | Intramuscular | LNP | CureVac AG | NCT04674189 | III |
| ARCT-021 | mRNA | Spike Glycoprotein | Intramuscular | LNP | Arcturus Therapeutics | NCT04668339 | II |
| LNP-nCoVsaRNA | mRNA | Spike Glycoprotein | Intramuscular | LNP | Imperial College London | ISRCTN17072692 | I |
| SARS-CoV-2 mRNA vaccine (ARCoV) | mRNA | RBD | Intramuscular | LNP | AMS/Walvax Biotechnology and Suzhou Abogen Biosciences | NCT04847102 | III |
| ChulaCov19 mRNA vaccine | mRNA | Spike Glycoprotein | Intramuscular | LNP | Chulalongkorn University | NCT04566276 | I |
| PTX-COVID19-B, mRNA vaccine | mRNA | Spike Glycoprotein | Intramuscular | LNP | Providence therapeutics | NCT04765436 | I |
| saRNA formulated in a NLC | mRNA | - | - | NLC | Infectious Disease Research Institute/Amyris, Inc. | - | Pre-Clinical |
| LNP-encapsulated mRNA encoding S | mRNA | Spike Glycoprotein | - | LNP | Max-Planck-Institute of Colloids and Interfaces | - | Pre-Clinical |
| Self-amplifying RNA | mRNA | - | - | - | Gennova | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | Selcuk University | - | Pre-Clinical |
| LNP-mRNA | mRNA | - | - | LNP | Translate Bio/Sanofi Pasteur | - | Pre-Clinical |
| LNP-mRNA | mRNA | - | - | LNP | CanSino Biologics/Precision NanoSystems | - | Pre-Clinical |
| LNP-encapsulated mRNA cocktail encoding VLP | mRNA | - | - | LNP | Fudan University/Shanghai JiaoTong University/RNACure Biopharma | - | Pre-Clinical |
| LNP-encapsulated mRNA encoding RBD | mRNA | RBD | - | LNP | Fudan University/Shanghai JiaoTong University/RNACure Biopharma | - | Pre-Clinical |
| Replicating Defective SARS-CoV-2 derived RNAs | mRNA | - | - | - | Centro Nacional Biotecnología (CNB-CSIC), Spain | - | Pre-Clinical |
| LNP-encapsulated mRNA | mRNA | - | - | LNP | University of Tokyo/Daiichi-Sankyo | - | Pre-Clinical |
| Liposome-encapsulated mRNA | mRNA | - | - | LNP | BIOCAD | - | Pre-Clinical |
| Several mRNA candidates | mRNA | - | - | - | RNAimmune, Inc. | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | FBRI SRC VB VECTOR, Rospotrebnadzor, Koltsovo | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | China CDC/Tongji University/Stermina | - | Pre-Clinical |
| mRNA in an intranasal delivery system | mRNA | - | Intranasal | - | eTheRNA | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | Greenlight Biosciences | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | IDIBAPS-Hospital Clinic, Spain | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | Providence Therapeutics | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | Cell Tech Pharmed | - | Pre-Clinical |
| mRNA | mRNA | - | - | - | ReNAP Co. | - | Pre-Clinical |
| D614G variant LNP-encapsulated mRNA | mRNA | - | - | LNP | Globe Biotech Ltd. | - | Pre-Clinical |
| Encapsulated mRNA | mRNA | - | - | - | CEA | - | Pre-Clinical |
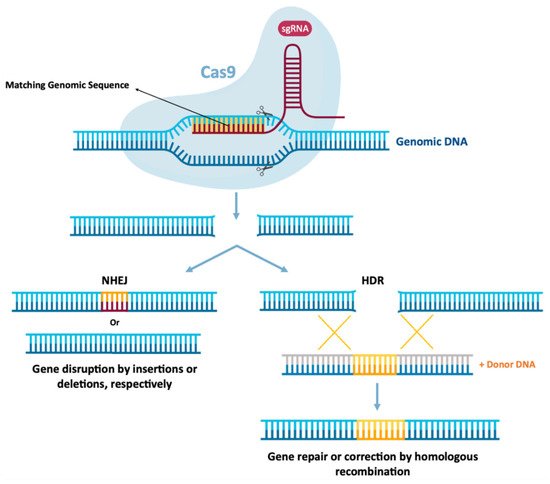
3. Gene Editing
This entry is adapted from the peer-reviewed paper 10.3390/pharmaceutics13122090
References
- Hajj, K.A.; Whitehead, K.A. Tools for translation: Non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2017, 2.
- Kowalski, P.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728.
- Sahu, I.; Haque, A.K.M.A.; Weidensee, B.; Weinmann, P.; Kormann, M.S.D. Recent Developments in mRNA-Based Protein Supplementation Therapy to Target Lung Diseases. Mol. Ther. 2019, 27, 803–823.
- Midoux, P.; Pichon, C. Lipid-based mRNA vaccine delivery systems. Expert Rev. Vaccines 2014, 14, 221–234.
- Gómez-Aguado, I.; Rodríguez-Castejón, J.; Vicente-Pascual, M.; Rodríguez-Gascón, A.; Aspiazu, M.; Ángeles, S.; Del Pozo-Rodríguez, A. Nanomedicines to Deliver mRNA: State of the Art and Future Perspectives. Nanomaterials 2020, 10, 364.
- Ramaswamy, S.; Tonnu, N.; Tachikawa, K.; Limphong, P.; Vega, J.B.; Karmali, P.P.; Chivukula, P.; Verma, I.M. Systemic delivery of factor IX messenger RNA for protein replacement therapy. Proc. Natl. Acad. Sci. USA 2017, 114, E1941–E1950.
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics—developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780.
- An, D.; Frassetto, A.; Jacquinet, E.; Eybye, M.; Milano, J.; DeAntonis, C.; Nguyen, V.; Laureano, R.; Milton, J.; Sabnis, S.; et al. Long-term efficacy and safety of mRNA therapy in two murine models of methylmalonic acidemia. EBioMedicine 2019, 45, 519–528.
- Baba, M.; Itaka, K.; Kondo, K.; Yamasoba, T.; Kataoka, K. Treatment of neurological disorders by introducing mRNA in vivo using polyplex nanomicelles. J. Control. Release 2015, 201, 41–48.
- Magadum, A.; Singh, N.; Kurian, A.A.; Sharkar, M.T.K.; Chepurko, E.; Zangi, L. Ablation of a Single N-Glycosylation Site in Human FSTL 1 Induces Cardiomyocyte Proliferation and Cardiac Regeneration. Mol. Ther.-Nucleic Acids 2018, 13, 133–143.
- Magadum, A.; Singh, N.; Kurian, A.A.; Munir, I.; Mehmood, T.; Brown, K.; Sharkar, M.T.K.; Chepurko, E.; Sassi, Y.; Oh, J.G.; et al. Pkm2 Regulates Cardiomyocyte Cell Cycle and Promotes Cardiac Regeneration. Circulation 2020, 141, 1249–1265.
- Lei, S.; Zhang, X.; Men, K.; Gao, Y.; Yang, X.; Wu, S.; Duan, X.; Wei, Y.; Tong, R. Efficient Colorectal Cancer Gene Therapy with IL-15 mRNA Nanoformulation. Mol. Pharm. 2020, 17.
- Jiang, T.; Zhou, C.; Ren, S. Role of IL-2 in cancer immunotherapy. OncoImmunology 2016, 5, e1163462.
- Beck, J.D.; Reidenbach, D.; Salomon, N.; Sahin, U.; Türeci, Ö.; Vormehr, M.; Kranz, L.M. mRNA therapeutics in cancer immunotherapy. Mol. Cancer 2021, 20, 69.
- Maruggi, G.; Zhang, C.; Li, J.; Ulmer, J.B.; Yu, D. mRNA as a Transformative Technology for Vaccine Development to Control Infectious Diseases. Mol. Ther. 2019, 27, 757–772.
- Schlake, T.; Thran, M.; Fiedler, K.; Heidenreich, R.; Petsch, B.; Fotin-Mleczek, M. mRNA: A Novel Avenue to Antibody Therapy? Mol. Ther. 2019, 27, 773–784.
- Delehedde, C.; Even, L.; Midoux, P.; Pichon, C.; Perche, F. Intracellular Routing and Recognition of Lipid-Based mRNA Nanoparticles. Pharmaceutics 2021, 13, 945.
- Shih, H.-I.; Wu, C.-J.; Tu, Y.-F.; Chi, C.-Y. Fighting COVID-19: A quick review of diagnoses, therapies, and vaccines. Biomed. J. 2020, 43, 341–354.
- Conry, R.M.; LoBuglio, A.F.; Wright, M.; Sumerel, L.; Pike, M.J.; Johanning, F.; Benjamin, R.; Lu, D.; Curiel, D.T. Characterization of a messenger RNA polynucleotide vaccine vector. Cancer Res. 1995, 55, 1397–1400.
- Billingsley, M.M.; Singh, N.; Ravikumar, P.; Zhang, R.; June, C.H.; Mitchell, M.J. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020, 20, 1578–1589.
- Granot-Matok, Y.; Kon, E.; Dammes, N.; Mechtinger, G.; Peer, D. Therapeutic mRNA delivery to leukocytes. J. Control. Release 2019, 305, 165–175.
- Belete, T.M. A review on Promising vaccine development progress for COVID-19 disease. Vacunas 2020, 21, 121–128.
- He, W.; Evans, A.C.; Rasley, A.; Bourguet, F.; Peters, S.; Kamrud, K.I.; Wang, N.; Hubby, B.; Felderman, M.; Gouvis, H.; et al. Cationic HDL mimetics enhance in vivo delivery of self-replicating mRNA. Nanomed. Nanotechnol. Biol. Med. 2020, 24, 102154.
- Versteeg, L.; Almutairi, M.M.; Hotez, P.J.; Pollet, J. Enlisting the mRNA Vaccine Platform to Combat Parasitic Infections. Vaccines 2019, 7, 122.
- Alton, E.W.F.W.; Armstrong, D.K.; Ashby, D.; Bayfield, K.J.; Bilton, D.; Bloomfield, E.V.; Boyd, A.C.; Brand, J.; Buchan, R.; Calcedo, R.; et al. Repeated nebulisation of non-viral CFTR gene therapy in patients with cystic fibrosis: A randomised, double-blind, placebo-controlled, phase 2b trial. Lancet Respir. Med. 2015, 3, 684–691.
- Trepotec, Z.; Lichtenegger, E.; Plank, C.; Aneja, M.K.; Rudolph, C. Delivery of mRNA Therapeutics for the Treatment of Hepatic Diseases. Mol. Ther. 2019, 27, 794–802.
- Alberer, M.; Gnad-Vogt, U.; Hong, H.S.; Mehr, K.T.; Backert, L.; Finak, G.; Gottardo, R.; Bica, M.A.; Garofano, A.; Koch, S.D.; et al. Safety and immunogenicity of a mRNA rabies vaccine in healthy adults: An open-label, non-randomised, prospective, first-in-human phase 1 clinical trial. Lancet 2017, 390, 1511–1520.
- Fotin-Mleczek, M.; Duchardt, K.M.; Lorenz, C.; Pfeiffer, R.; Ojkić-Zrna, S.; Probst, J.; Kallen, K.-J. Messenger RNA-based Vaccines with Dual Activity Induce Balanced TLR-7 Dependent Adaptive Immune Responses and Provide Antitumor Activity. J. Immunother. 2011, 34, 1–15.
- Feldman, R.A.; Fuhr, R.; Smolenov, I.; Ribeiro, A.; Panther, L.; Watson, M.; Senn, J.J.; Smith, M.; Almarsson, Ӧ.; Pujar, H.S.; et al. mRNA vaccines against H10N8 and H7N9 influenza viruses of pandemic potential are immunogenic and well tolerated in healthy adults in phase 1 randomized clinical trials. Vaccine 2019, 37, 3326–3334.
- Bos, R.; Marquardt, K.L.; Cheung, J.; Sherman, L.A. Functional differences between low- and high-affinity CD8+ T cells in the tumor environment. OncoImmunology 2012, 1, 1239–1247.
- Klein, L.; Hinterberger, M.; Wirnsberger, G.; Kyewski, B. Antigen presentation in the thymus for positive selection and central tolerance induction. Nat. Rev. Immunol. 2009, 9, 833–844.
- Zhong, Z.; Mc Cafferty, S.; Combes, F.; Huysmans, H.; De Temmerman, J.; Gitsels, A.; Vanrompay, D.; Catani, J.P.; Sanders, N.N. mRNA therapeutics deliver a hopeful message. Nano Today 2018, 23, 16–39.
- Leal, L.; Guardo, A.C.; Morón-López, S.; Salgado, M.; Mothe, B.; Heirman, C.; Pannus, P.; Vanham, G.; van den Ham, H.J.; Gruters, R.; et al. Phase I clinical trial of an intranodally administered mRNA-based therapeutic vaccine against HIV-1 infection. AIDS 2018, 32, 2533–2545.
- De Jong, W.; Aerts, J.; Allard, S.; Brander, C.; Buyze, J.; Florence, E.; van Gorp, E.; Vanham, G.; Leal, L.; Mothe, B. iHIVARNA phase IIa, a randomized, placebo-controlled, double-blinded trial to evaluate the safety and immunogenicity of iHIVARNA-01 in chronically HIV-infected patients under stable combined antiretroviral therapy. Trials 2019, 20, 1–10.
- Gandhi, R.T.; Kwon, D.S.; Macklin, E.A.; Shopis, J.R.; McLean, A.P.; McBrine, N.; Flynn, T.; Peter, L.; Sbrolla, A.; Kaufmann, D.E. Immunization of HIV-1-infected persons with autologous dendritic cells transfected with mRNA encoding HIV-1 Gag and Nef: Results of a randomized, placebo-controlled clinical trial. J. Acquir. Immune Defic. Syndr. 2016, 71, 246.
- Kyte, J.A.; Mu, L.; Aamdal, S.; Kvalheim, G.; Dueland, S.; Hauser, M.; Gullestad, H.P.; Ryder, T.; Lislerud, K.; Hammerstad, H.; et al. Phase I/II trial of melanoma therapy with dendritic cells transfected with autologous tumor-mRNA. Cancer Gene Ther. 2006, 13, 905–918.
- Wecker, M.; Gilbert, P.; Russell, N.; Hural, J.; Allen, M.; Pensiero, M.; Chulay, J.; Chiu, Y.-L.; Karim, S.S.A.; Burke, D.S. Phase I Safety and Immunogenicity Evaluations of an Alphavirus Replicon HIV-1 Subtype C gag Vaccine in Healthy HIV-1-Uninfected Adults. Clin. Vaccine Immunol. 2012, 19, 1651–1660.
- Hu, Z.; Ding, W.; Zhu, D.; Yu, L.; Jiang, X.; Wang, X.; Zhang, C.; Wang, L.; Ji, T.; Liu, D.; et al. TALEN-mediated targeting of HPV oncogenes ameliorates HPV-related cervical malignancy. J. Clin. Investig. 2014, 125, 425–436.
- Hu, Z.; Yu, L.; Zhu, D.; Ding, W.; Wang, X.; Zhang, C.; Wang, L.; Jiang, X.; Shen, H.; He, D.; et al. Disruption of HPV16-E7 by CRISPR/Cas System Induces Apoptosis and Growth Inhibition in HPV16 Positive Human Cervical Cancer Cells. BioMed Res. Int. 2014, 2014, 1–9.
- Qasim, W.; Zhan, H.; Samarasinghe, S.; Adams, S.; Amrolia, P.; Stafford, S.; Butler, K.; Rivat, C.; Wright, G.; Somana, K.; et al. Molecular remission of infant B-ALL after infusion of universal TALEN gene-edited CAR T cells. Sci. Transl. Med. 2017, 9, eaaj2013.
- Su, S.; Hu, B.; Shao, J.; Shen, B.; Du, J.; Du, Y.; Zhou, J.; Yu, L.; Zhang, L.; Chen, F.; et al. CRISPR-Cas9 mediated efficient PD-1 disruption on human primary T cells from cancer patients. Sci. Rep. 2016, 6, 20070.
- Quan, L.; Chen, X.; Liu, A.; Zhang, Y.; Guo, X.; Yan, S.; Liu, Y. PD-1 Blockade Can Restore Functions of T-Cells in Epstein-Barr Virus-Positive Diffuse Large B-Cell Lymphoma In Vitro. PLoS ONE 2015, 10, e0136476.
- DiGiusto, D.L.; Cannon, P.M.; Holmes, M.C.; Li, L.; Rao, A.; Wang, J.; Lee, G.; Gregory, P.; Kim, K.A.; Hayward, S.B.; et al. Preclinical development and qualification of ZFN-mediated CCR5 disruption in human hematopoietic stem/progenitor cells. Mol. Ther.—Methods Clin. Dev. 2016, 3, 16067.
- Samson, M.; Libert, F.; Doranz, B.J.; Rucker, J.; Liesnard, C.; Farber, C.-M.; Saragosti, S.; Lapoumeroulie, C.; Cognaux, J.; Forceille, C.; et al. Resistance to HIV-1 infection in Caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 1996, 382, 722–725.
- Ahammad, I.; Lira, S.S. Designing a novel mRNA vaccine against SARS-CoV-2: An immunoinformatics approach. Int. J. Biol. Macromol. 2020, 162, 820–837.
- Khuroo, M.S.; Khuroo, M.; Khuroo, M.S.; Sofi, A.A.; Khuroo, N.S. COVID-19 Vaccines: A Race Against Time in the Middle of Death and Devastation! J. Clin. Exp. Hepatol. 2020, 10, 610–621.
- Velavan, T.P.; Meyer, C.G. The COVID-19 epidemic. Trop. Med. Int. Health 2020, 25, 278–280.
- Yang, P.; Wang, X. COVID-19: A new challenge for human beings. Cell. Mol. Immunol. 2020, 17, 555–557.
- Ahmed, S.F.; Quadeer, A.A.; McKay, M.R. Preliminary identification of potential vaccine targets for the COVID-19 coronavirus (SARS-CoV-2) based on SARS-CoV immunological studies. Viruses 2020, 12, 254.
- Koirala, A.; Joo, Y.J.; Khatami, A.; Chiu, C.; Britton, P.N. Vaccines for COVID-19: The current state of play. Paediatr. Respir. Rev. 2020, 35, 43–49.
- Caddy, S. Developing a vaccine for COVID-19. BMJ 2020, 369, m1790.
- Yang, N.; Shen, H.-M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724–1731.
- Liu, C.; Zhou, Q.; Li, Y.; Garner, L.V.; Watkins, S.; Carter, L.J.; Smoot, J.; Gregg, A.C.; Daniels, A.D.; Jervey, S.; et al. Research and Development on Therapeutic Agents and Vaccines for COVID-19 and Related Human Coronavirus Diseases. ACS Cent. Sci. 2020, 6, 315–331.
- Lurie, N.; Saville, M.; Hatchett, R.; Halton, J. Developing COVID-19 Vaccines at Pandemic Speed. N. Engl. J. Med. 2020, 382, 1969–1973.
- Thanh Le, T.; Andreadakis, Z.; Kumar, A.; Gómez Román, R.; Tollefsen, S.; Saville, M.; Mayhew, S. The COVID-19 vaccine development landscape. Nat. Rev. Drug Discov. 2020, 19, 305–306.
- Corey, B.L.; Mascola, J.R.; Fauci, A.S.; Collins, F.S. A strategic approach to COVID-19 vaccine R&D. Science 2020, 368, 948–950.
- Kowalzik, F.; Schreiner, D.; Jensen, C.; Teschner, D.; Gehring, S.; Zepp, F. mRNA-Based Vaccines. Vaccines 2021, 9, 390.
- Oberhardt, V.; Luxenburger, H.; Kemming, J.; Schulien, I.; Ciminski, K.; Giese, S.; Csernalabics, B.; Lang-Meli, J.; Janowska, I.; Staniek, J.; et al. Rapid and stable mobilization of CD8+ T cells by SARS-CoV-2 mRNA vaccine. Nature 2021, 597, 268–273.
- Voysey, M.; Clemens, S.A.C.; Madhi, S.A.; Weckx, L.Y.; Folegatti, P.M.; Aley, P.K.; Angus, B.; Baillie, V.L.; Barnabas, S.L.; Bhorat, Q.E.; et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: An interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. Lancet 2021, 397, 99–111.
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and Efficacy of the BNT162b2 mRNA COVID-19 Vaccine. N. Engl. J. Med. 2020, 383, 2603–2615.
- Kim, J.H.; Marks, F.; Clemens, J.D. Looking beyond COVID-19 vaccine phase 3 trials. Nat. Med. 2021, 27, 205–211.
- Baden, L.R.; El Sahly, H.M.; Essink, B.; Kotloff, K.; Frey, S.; Novak, R.; Diemert, D.; Spector, S.A.; Rouphael, N.; Creech, C.B. Efficacy and safety of the mRNA-1273 SARS-CoV-2 vaccine. N. Engl. J. Med. 2021, 384, 403–416.
- Comirnaty Epar Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/comirnaty-epar-product-information_en.pdf (accessed on 18 November 2021).
- Spikevax previously COVID-19 vaccine Moderna Epar Product Information. Available online: https://www.ema.europa.eu/en/documents/product-information/spikevax-previously-covid-19-vaccine-moderna-epar-product-information_en.pdf (accessed on 18 November 2021).
- Lamb, Y.N. BNT162b2 mRNA COVID-19 Vaccine: First Approval. Drugs 2021, 81, 495–501.
- Fact Sheet for Healthcare Providers Administering Vaccine (Vaccination Providers)—Emergency Use Authorization (EUA) of the Pfizer-Biontech COVID-19 Vaccine to Prevent Coronavirus Disease 2019 (COVID-19). Available online: https://www.fda.gov/media/153713/download (accessed on 26 November 2021).
- Fact Sheet for Healthcare Providers Administering Vaccine. Moderna. U.S. Food Drug Adm. 2020, 8, 55. Available online: https://www.fda.gov/media/144637/download (accessed on 8 May 2021).
- Comirnaty (COVID-19 mRNA Vaccine )—An Overview of Comirnaty and Why It Is Authorised in the EU. Available online: https://www.ema.europa.eu/en/documents/overview/comirnaty-epar-medicine-overview_en.pdf (accessed on 26 November 2021).
- Spikevax (COVID-19 mRNA Vaccine )—An Overview of Spikevax and Why It Is Authorised in the EU. Available online: https://www.ema.europa.eu/en/documents/overview/spikevax-previously-covid-19-vaccine-moderna-epar-medicine-overview_en.pdf (accessed on 26 November 2021).
- Widge, A.T.; Rouphael, N.G.; Jackson, L.A.; Anderson, E.J.; Roberts, P.C.; Makhene, M.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; Pruijssers, A.J.; et al. Durability of Responses after SARS-CoV-2 mRNA-1273 Vaccination. N. Engl. J. Med. 2020, 384, 80–82.
- Meo, S.A.; Bukhari, I.A.; Akram, J.; Meo, A.S.; Klonoff, D.C. COVID-19 vaccines: Comparison of biological, pharmacological characteristics and adverse effects of Pfizer/BioNTech and Moderna Vaccines. Eur. Rev. Med. Pharmacol. Sci. 2021, 25, 1663–1669.
- Kremsner, P.G.; Mann, P.; Kroidl, A.; Leroux-Roels, I.; Schindler, C.; Gabor, J.J.; Schunk, M.; Leroux-Roels, G.; Bosch, J.J.; Fendel, R.; et al. Safety and immunogenicity of an mRNA-lipid nanoparticle vaccine candidate against SARS-CoV-2. Wien. Klin. Wochenschr. 2021, 133, 931–941.
- Rauch, S.; Roth, N.; Schwendt, K.; Fotin-Mleczek, M.; Mueller, S.O.; Petsch, B. mRNA-based SARS-CoV-2 vaccine candidate CVnCoV induces high levels of virus-neutralising antibodies and mediates protection in rodents. npj Vaccines 2021, 6, 57.
- Karikó, K.; Buckstein, M.; Ni, H.; Weissman, D. Suppression of RNA Recognition by Toll-like Receptors: The Impact of Nucleoside Modification and the Evolutionary Origin of RNA. Immunity 2005, 23, 165–175.
- Abu-Raddad, L.J.; Chemaitelly, H.; Butt, A.A. Effectiveness of the BNT162b2 COVID-19 Vaccine against the B.1.1.7 and B.1.351 Variants. N. Engl. J. Med. 2021, 385, 187–189.
- Sheikh, A.; McMenamin, J.; Taylor, B.; Robertson, C.; Public Health Scotland and the EAVE II Collaborators. SARS-CoV-2 Delta VOC in Scotland: Demographics, risk of hospital admission, and vaccine effectiveness. Lancet 2021, 397, 2461–2462.
- Oliver, S.E.; Gargano, J.W.; Marin, M.; Wallace, M.; Curran, K.G.; Chamberland, M.; McClung, N.; Campos-Outcalt, D.; Morgan, R.L.; Mbaeyi, S.; et al. The Advisory Committee on Immunization Practices’ Interim Recommendation for Use of Moderna COVID-19 Vaccine - United States, December 2020. MMWR Morb. Mortal. Wkly. Rep. 2021, 69, 1653–1656.
- Vaidyanathan, S.; Azizian, K.T.; Haque, A.A.; Henderson, J.M.; Hendel, A.; Shore, S.; Antony, J.S.; Hogrefe, R.I.; Kormann, M.S.; Porteus, M.H.; et al. Uridine Depletion and Chemical Modification Increase Cas9 mRNA Activity and Reduce Immunogenicity without HPLC Purification. Mol. Ther.—Nucleic Acids 2018, 12, 530–542.
- Karikó, K.; Muramatsu, H.; Welsh, F.A.; Ludwig, J.; Kato, H.; Akira, S.; Weissman, D. Incorporation of Pseudouridine Into mRNA Yields Superior Nonimmunogenic Vector with Increased Translational Capacity and Biological Stability. Mol. Ther. 2008, 16, 1833–1840.
- Eyler, D.E.; Franco, M.K.; Batool, Z.; Wu, M.Z.; Dubuke, M.L.; Dobosz-Bartoszek, M.; Jones, J.; Polikanov, Y.S.; Roy, B.; Koutmou, K.S. Pseudouridinylation of mRNA coding sequences alters translation. Proc. Natl. Acad. Sci. USA 2019, 116, 23068–23074.
- Nance, K.D.; Meier, J.L. Modifications in an Emergency: The Role of N1-Methylpseudouridine in COVID-19 Vaccines. ACS Cent. Sci. 2021, 7, 748–756.
- Alexaki, A.; Hettiarachchi, G.K.; Athey, J.C.; Katneni, U.; Simhadri, V.; Hamasaki-Katagiri, N.; Nanavaty, P.; Lin, B.; Takeda, K.; Freedberg, D.; et al. Effects of codon optimization on coagulation factor IX translation and structure: Implications for protein and gene therapies. Sci. Rep. 2019, 9, 1–15.
- Holtkamp, S.; Kreiter, S.; Selmi, A.; Simon, P.; Koslowski, M.; Huber, C.; Türeci, O.; Sahin, U. Modification of antigen-encoding RNA increases stability, translational efficacy, and T-cell stimulatory capacity of dendritic cells. Blood 2006, 108, 4009–4017.
- OMS. COVID-19 Vaccine Tracker and Landscape. Available online: https://www.who.int/publications/m/item/draft-landscape-of-covid-19-candidate-vaccines (accessed on 1 October 2021).
- Ghosh, D.; Venkataramani, P.; Nandi, S.; Bhattacharjee, S. CRISPR–Cas9 a boon or bane: The bumpy road ahead to cancer therapeutics. Cancer Cell Int. 2019, 19, 1–10.
- Janssen, J.M.; Chen, X.; Liu, J.; Gonçalves, M.A. The Chromatin Structure of CRISPR-Cas9 Target DNA Controls the Balance between Mutagenic and Homology-Directed Gene-Editing Events. Mol. Ther.-Nucleic Acids 2019, 16, 141–154.
- Anzalone, A.V.; Randolph, P.B.; Davis, J.R.; Sousa, A.A.; Koblan, L.W.; Levy, J.M.; Chen, P.J.; Wilson, C.; Newby, G.A.; Raguram, A.; et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 2019, 576, 149–157.
- Shen, B.; Zhang, W.; Zhang, J.; Zhou, J.; Wang, J.; Chen, L.; Wang, L.; Hodgkins, A.; Iyer, V.; Huang, X.; et al. Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat. Methods 2014, 11, 399–402.
- Miller, J.B.; Zhang, S.; Kos, P.; Xiong, H.; Zhou, K.; Perelman, S.S.; Zhu, H.; Siegwart, D.J. Non-Viral CRISPR/Cas Gene Editing In Vitro and In Vivo Enabled by Synthetic Nanoparticle Co-Delivery of Cas9 mRNA and sgRNA. Angew. Chem. Int. Ed. Engl. 2017, 56, 1059–1063.
- Liang, X.; Potter, J.; Kumar, S.; Zou, Y.; Quintanilla, R.; Sridharan, M.; Carte, J.; Chen, W.; Roark, N.; Ranganathan, S.; et al. Rapid and highly efficient mammalian cell engineering via Cas9 protein transfection. J. Biotechnol. 2015, 208, 44–53.
- Mohsin, M.; Li, Y.; Zhang, X.; Wang, Y.; Huang, Z.; Yin, G.; Zhang, Z. Development of CRISPR-CAS9 based RNA drugs against Eimeria tenella infection. Genomics 2021, 113, 4126–4135.
- Gratz, S.J.; Cummings, A.M.; Nguyen, J.N.; Hamm, D.C.; Donohue, L.K.; Harrison, M.M.; Wildonger, J.; O’Connor-Giles, K.M. Genome Engineering of Drosophila with the CRISPR RNA-Guided Cas9 Nuclease. Genetics 2013, 194, 1029–1035.
- Wang, H.; Yang, H.; Shivalila, C.S.; Dawlaty, M.M.; Cheng, A.W.; Zhang, F.; Jaenisch, R. One-Step Generation of Mice Carrying Mutations in Multiple Genes by CRISPR/Cas-Mediated Genome Engineering. Cell 2013, 153, 910–918.
- Hwang, W.Y.; Fu, Y.; Reyon, D.; Maeder, M.L.; Tsai, S.Q.; Sander, J.D.; Peterson, R.T.; Yeh, J.-R.J.; Joung, J.K. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nat. Biotechnol. 2013, 31, 227–229.
- Niu, Y.; Shen, B.; Cui, Y.; Chen, Y.; Wang, J.; Wang, L.; Kang, Y.; Zhao, X.; Si, W.; Li, W.; et al. Generation of Gene-Modified Cynomolgus Monkey via Cas9/RNA-Mediated Gene Targeting in One-Cell Embryos. Cell 2014, 156, 836–843.
- Ling, S.; Yang, S.; Hu, X.; Yin, D.; Dai, Y.; Qian, X.; Wang, D.; Pan, X.; Hong, J.; Sun, X.; et al. Lentiviral delivery of co-packaged Cas9 mRNA and a Vegfa-targeting guide RNA prevents wet age-related macular degeneration in mice. Nat. Biomed. Eng. 2021, 5, 144–156.
- Qiu, M.; Glass, Z.; Chen, J.; Haas, M.; Jin, X.; Zhao, X.; Rui, X.; Ye, Z.; Li, Y.; Zhang, F.; et al. Lipid nanoparticle-mediated codelivery of Cas9 mRNA and single-guide RNA achieves liver-specific in vivo genome editing of Angptl3. Proc. Natl. Acad. Sci. USA 2021, 118.
- Graham, M.J.; Lee, R.G.; Brandt, T.A.; Tai, L.-J.; Fu, W.; Peralta, R.; Yu, R.; Hurh, E.; Paz, E.; McEvoy, B.W.; et al. Cardiovascular and Metabolic Effects of ANGPTL3 Antisense Oligonucleotides. N. Engl. J. Med. 2017, 377, 222–232.
- Abbott, T.R.; Dhamdhere, G.; Liu, Y.; Lin, X.; Goudy, L.; Zeng, L.; Chemparathy, A.; Chmura, S.; Heaton, N.S.; Debs, R.; et al. Development of CRISPR as an Antiviral Strategy to Combat SARS-CoV-2 and Influenza. Cell 2020, 181, 865.e12–876.e12.
- Guo, X.; Rahman, J.A.; Wessels, H.-H.; Méndez-Mancilla, A.; Haro, D.; Chen, X.; Sanjana, N.E. Transcriptome-wide Cas13 guide RNA design for model organisms and viral RNA pathogens. Cell Genom. 2021, 1, 100001.
- Kushawah, G.; Hernandez-Huertas, L.; Abugattas-Nuñez Del Prado, J.; Martinez-Morales, J.R.; DeVore, M.L.; Hassan, H.; Moreno-Sanchez, I.; Tomas-Gallardo, L.; Diaz-Moscoso, A.; Monges, D.E.; et al. CRISPR-Cas13d Induces Efficient mRNA Knockdown in Animal Embryos. Dev. Cell 2020, 54, 805.e7–817.e7.
- Xu, D.; Cai, Y.; Tang, L.; Han, X.; Gao, F.; Cao, H.; Qi, F.; Kapranov, P. A CRISPR/Cas13-based approach demonstrates biological relevance of vlinc class of long non-coding RNAs in anticancer drug response. Sci. Rep. 2020, 10, 1–13.
- Patchsung, M.; Jantarug, K.; Pattama, A.; Aphicho, K.; Suraritdechachai, S.; Meesawat, P.; Sappakhaw, K.; Leelahakorn, N.; Ruenkam, T.; Wongsatit, T.; et al. Clinical validation of a Cas13-based assay for the detection of SARS-CoV-2 RNA. Nat. Biomed. Eng. 2020, 4, 1140–1149.
- Blanchard, E.L.; Vanover, D.; Bawage, S.S.; Tiwari, P.M.; Rotolo, L.; Beyersdorf, J.; Peck, H.E.; Bruno, N.C.; Hincapie, R.; Michel, F.; et al. Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Nat. Biotechnol. 2021, 39, 717–726.
- Rashnonejad, A.; Chermahini, G.A.; Wallace, L.; Harper, S.O. 8DUX4 mRNA silencing with CRISPR-Cas13 gene therapy as a prospective treatment for Facioscapulohumeral muscular dystrophy. Neuromuscul. Disord. 2019, 29, S40.