Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Biochemistry & Molecular Biology
For eukaryotic cells, reactive oxygen species (ROS) encompass a group of molecules derived from oxygen. Due to the well-established role of ROS in cell signaling, cancer cells always have higher levels of endogenous ROS to enhance rapid cell growth and proliferation through the mitogen-activated protein kinase (MAPK)/extracellular-regulated kinase 1/2 (ERK1/2), phosphoinositide-3-kinase (PI3K)/Akt, nuclear factor-κB (NF-κB), and hypoxia-sensitive α (HIF1α) pathways.
- anti-cancer
- reactive oxygen species
- chelators
- metal complexes
- antioxidant enzymes
- SOD1
- TrxR
- mitochondria
1. Introduction
For eukaryotic cells, reactive oxygen species (ROS) encompass a group of molecules derived from oxygen, such as hydrogen peroxide (H2O2), superoxide anion (O2•−), organic hydroperoxides (ROOH), singlet molecular oxygen (1O2), hydroxyl radical (•OH), alkoxyl radical (•OR), and peroxyl radical (•OOR) [1,2,3]. ROS are mainly formed by reduction–oxidation reactions or by electronic excitation (Figure 1A) [1,2,3] and have evolved as regulators of multiple signaling pathways [4,5,6,7,8]. Two species, H2O2 and O2•−, are the most important redox signaling agents in the cells [4,5,6,7,8]. H2O2 is the major ROS in organisms, with its concentration always maintained within 1~100 nM under normal conditions [8]. For O2•−, the concentration is also maintained at about 0.01 nM, much lower than that of H2O2 [8].
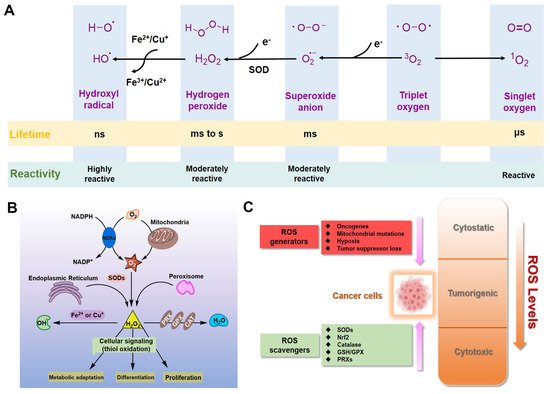
Figure 1. ROS in cancer cells. (A) Generation and chemical structures of ROS. (B) Brief metabolic process and signal regulation of intracellular ROS. (C) Balancing ROS generation and scavenging in cancer cells to remain in the tumorigenic range.
Dozens of growth factors, cytokines, and antioxidant enzymes control the homeostasis of intracellular H2O2 and O2•− (Figure 1B) [9]. O2•− is prominently generated by the mitochondrial electron transport chain (Mito-ETC), NADPH oxidase (NOX) complex, and endoplasmic reticulum (ER) system, and is rapidly converted to H2O2 by superoxide dismutases (SODs) [10,11,12]. Subsequently, H2O2 is mainly detoxified to H2O by catalase (CAT), glutathione peroxidase (GPX), and peroxiredoxin (Prx) [12]. It is worth mentioning that •OH formed by metal-catalyzed Fenton reaction is the most reactive ROS; it can oxidize biological macromolecules indiscriminately, such as DNA, proteins, and lipids [12]. Therefore, maintaining the homeostasis of intracellular ROS is essential for cell growth, proliferation, and survival [6,9,10].
Due to the well-established role of ROS in cell signaling, cancer cells always have higher levels of endogenous ROS to enhance rapid cell growth and proliferation through the mitogen-activated protein kinase (MAPK)/extracellular-regulated kinase 1/2 (ERK1/2), phosphoinositide-3-kinase (PI3K)/Akt, nuclear factor-κB (NF-κB), and hypoxia-sensitive α (HIF1α) pathways [13,14,15,16,17,18]. Indeed, higher levels of ROS have already been observed in various cancer cells [11,19]. If the intracellular ROS levels increase dramatically to toxic concentrations, oxidative stress will cause irreversible damage and may eventually lead to the death of cancer cells [11,20]. To maintain the elevated mitogenic signaling without incurring substantial oxidative damage by a proper balance of ROS, the antioxidant enzymes in cancer cells, such as Cu/Zn superoxide dismutase (SOD1), GPX, and Prx, should harbor higher levels of activity (Figure 1C) [21,22,23,24].
Elevated levels of ROS are always involved in the initiation and progression of cancer. Hence, intervening in the homeostasis of ROS in cancer cells is an effective anti-cancer strategy [25,26]. So far, a variety of chelators or metal complexes based on the regulation of ROS have been reported as anti-cancer agents [27,28,29,30,31,32,33,34]. For those antioxidant enzymes where the active center is a metal ion, chelators can be used to competitively bind to the metal ion and thus inhibit the enzymatic activity to achieve the regulation of intracellular ROS, including SOD1 inhibitors tetrathiomolybdate (ATN-224) and LD100 [32,33]. On the other hand, several metal complexes can regulate the ROS levels in cancer cells through other mechanisms to achieve anti-cancer purposes, such as TxrR inhibition and mitochondrial dysfunction [34,35,36]. Regulating the relative level of ROS in cancer cells through the mechanism of metal coordination has become an important branch with broad prospects in the field of cancer therapy.
2. Inorganic SOD1 Inhibitors with Anti-Cancer Prospects
In mammals, the main biological function of SODs is to catalyze the dismutation of O2•− into H2O2 and O2 [39,40]. Cu/Zn superoxide dismutase (SOD1), the major SOD, mainly exists in the formation of homodimers in cells and is widely distributed in the nucleus, the cytoplasm, and the intermembrane space (IMS) of mitochondria [41]. Next, Mn superoxide dismutase (SOD2) exclusively exists in the mitochondrial matrix [42]. An extracellular form of SOD (EC-SOD), also a Cu/Zn-containing SOD, is tetrameric and exists in most mammals [42]. Besides this, SOD1 also regulates multiple redox signals to control growth and metabolic pathways, such as glucose metabolism and transcription [5,43,44,45]. Therefore, SODs, especially SOD1, are the first firewall to resist oxidative stress.
Recently, emerging evidence from researchers has indicated that SOD1 is usually overexpressed in cancer cells; its activity is essential to maintain higher ROS levels under the critical threshold during aberrant energy metabolism of cancer progression [41]. For example, SOD1 accumulations were observed not only in the cytoplasm but also in the nucleus of human primary breast and mammary cancers [46]. Besides this, prostate cancer cells (DU145) also have higher levels of activity and expression of SOD1, compared with normal prostate cells (RWPE-1) [5]. In vitro studies also showed that the fast growth of non-small cell lung cancer (NSCLC) and leukemia depends on the high activity of SOD1, which controls the oncogenic KRAS and EGFR pathways [47,48], as well as other cancer cells and xenograft tumors [49]. In general, SOD1 is recognized as a promising anti-cancer target, and several small-molecule targeting drugs for SOD1 have already entered the preclinical and clinical development stages [50].
Since the activity of SOD1 mainly comes from the copper ion in the active center, a vast majority of SOD1 inhibitors are competitive chelators of copper ions. In 1975, Heikkila et al. found that diethyldithiocarbamate (DDC) can competitively bind to copper ions (Figure 2), thereby inhibiting SOD1 activity at a millimolar level [51]. After being inhibited by DDC, SOD1 cannot restore enzyme activity through dialysis, but adding CuSO4 during dialysis restores SOD1 activity [51]. In 1979, Misra systematically explored the mechanism by which DDC inhibits SOD1 activity [52]. In Phase I, one DDC molecule first coordinates with the copper(II) center in SOD1, with retention of activity. In Phase II, a second DDC displaces the copper(II) center, with a loss of activity. The shortcomings of DDC as a SOD1 inhibitor are mainly reflected in its high working concentration and poor specificity, such as its interference with the activity of cytochrome c oxidase [53,54]. Nevertheless, DDC still has a wide range of anti-cancer applications, and DDC effectively inhibits SOD1 activity to kill cancer cells [55,56,57].
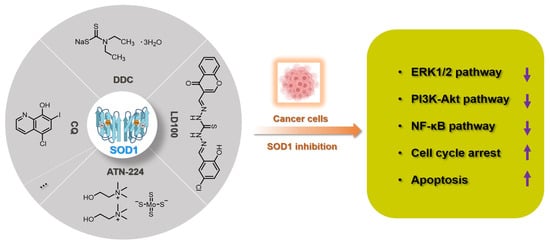
Figure 2. Summary of SOD1 inhibitors based on copper chelation. The specific activity inhibition of SOD1 selectively kills cancer cells by regulating the intracellular ROS signaling network.
In 2005, Ding et al. found that clioquinol (5-chloro-7-iodo-8-hydroxyquinoline, CQ), a metal chelator of copper/zinc/iron, is another SOD1 inhibitor (Figure 2), because the copper and zinc ions in the active sites of SOD1 are coordinated by CQ [58]. Structural characterization of the zinc(II) and copper(II) complexes with CQ indicated that the stoichiometry of ligand to metal is 2:1 [59]. Therefore, CQ can effectively inhibit SOD1 at micromolar concentrations (IC50: 6.7~43.1 μM) and induce the death of a variety of cancer cells through the caspase-3-mediated apoptosis pathway [58]. It cannot be ignored that CQ has the risk of destroying copper homeostasis during its inhibition of SOD1 activity in cells [60].
Tetrathiomolybdate is an orally available copper chelator that has been shown to have efficacy as an anti-angiogenic and anti-tumor agent in multiple cancers [61,62]. ATN-224 is the second-generation choline salt of tetrathiomolybdate with improved performance (Figure 2) and is being evaluated in several phase II trials in cancer patients [63]. Doñate et al. found that ATN-224 can selectively bind copper with high affinity, and SOD1 is the main target for the anti-angiogenic activity of this chelator [32,64]. ATN-224 also inhibits intracellular SOD1 activity at micromolar concentrations (IC50: 1.4~185 μM), but has specificity for copper binding, all of which makes it one of the most popular SOD1 inhibitors [62,64]. Every sulfur atom in tetrathiomolybdate can coordinate with copper and may then form metal clusters with copper enzymes, thereby inhibiting the activity of copper proteins, such as SOD1, cytochrome c oxidase, and ceruloplasmin [65,66]. Therefore, ATN-224 may also interfere with intracellular copper homeostasis or inhibit other copper enzymes. In cancer treatment, ATN-224-mediated SOD1 inhibition led to the downregulation of PDGF and increase of O2•−, prevented the formation of high levels of H2O2, and protected protein tyrosine phosphatases from oxidation by H2O2 [62]. Therefore, SOD1 inhibition by ATN-224 results in the down-regulation of multiple signaling pathways for cancer cell function, such as ERK1/2 and anti-apoptotic factor Mcl1 [50,62].
Considering that the known SOD1 inhibitors have various defects, such as low efficiency, weak specificity, and interference with the homeostasis of metal ions, we designed a next-generation SOD1 inhibitor (LD100) based on copper coordination chemistry and the catalytic cycle in the active site (Figure 2) [33]. LD100 was designed through the combination of thiosemicarbazone and phenol derivatives, because thiosemicarbazone contains a copper chelating moiety, -C(SH)-NH-, and the phenolic hydroxyl can further facilitate the copper coordination. Besides this, LD100 also contains a fluorescent group chromone, which not only can be used to track the entry of LD100 into cells, but also enables LD100 to better occupy the substrate channel of SOD1. Therefore, LD100 has a strong binding ability to copper ions in solution and can effectively inhibit the activity of SOD1 in vitro and in vivo (IC50 of LD100 to SOD1 in HeLa cells: 0.18 μM) [33]. Through specific inhibition of SOD1 activity, LD100 can efficiently up-regulate the intracellular concentration of O2•−, down-regulate the concentration of H2O2, down-regulate the phosphorylation of ERK1/2, and finally induce the apoptosis of cancer cells [33]. In summary, LD100 may be the most effective SOD1 inhibitor so far and has application prospects for cancer treatment. Using this inhibitor, we also systematically explored the mechanism of how SOD1 activity inhibition selectively kills cancer cells [5]. The rapid growth and proliferation of cancer cells always depend on higher SOD1 activity, so cancer cells are more sensitive to SOD1 inhibition. During SOD1 inhibition in cancer cells, LD100 could repress the ERK, PI3K-Akt, and NF-κB pathways; arrest the cell cycle; and induce mitochondria-dependent apoptosis [5].
SOD1 is indeed a recognized target for cancer treatment. At present, a variety of chelators have been used for SOD1 inhibition. LD100 may be the most effective inhibitor designed through coordination chemistry. However, the use of inorganic strategies to develop anti-cancer drugs based on SOD1 inhibition still requires further efforts. First, we need to solve the problem of compatibility between targeted and clinical deliveries. On the other hand, we also should reduce the side effects of chelating agents while ensuring the efficiency of SOD1 inhibition. The summary of SOD1 metal-chelating inhibition can provide a reference for the design of SOD1 inhibitors with anti-cancer effects in the future.
This entry is adapted from the peer-reviewed paper 10.3390/molecules27010148
This entry is offline, you can click here to edit this entry!