Cardiovascular disorders (CVDs) are the number one cause of death globally, according to the World Health Organization, being responsible for 17.9 million deaths in 2016, which represented 31% of all global deaths. Human pluripotent stem cells (hPSCs) have aroused attention as a powerful source of cardiac cells that could help to mitigate some of these problems, namely (1) the identification of new mechanisms of action in different cardiac disorders; (2) improving the reliability of cardiotoxicity side effect detection in newly developed compounds; and (3) providing a source of cardiac cells for the development of new regenerative medicine-based therapies.
- human pluripotent stem cells (hPSCs)
- hPSC-derived cardiac cells
- 3D cardiomyocyte (CM) differentiation
- 3D engineered cardiac microtissues (MTs)
- engineered heart tissues (EHT)
- drug screening platforms
- cardiotoxicity
- cardiac disease modeling
1. Introduction
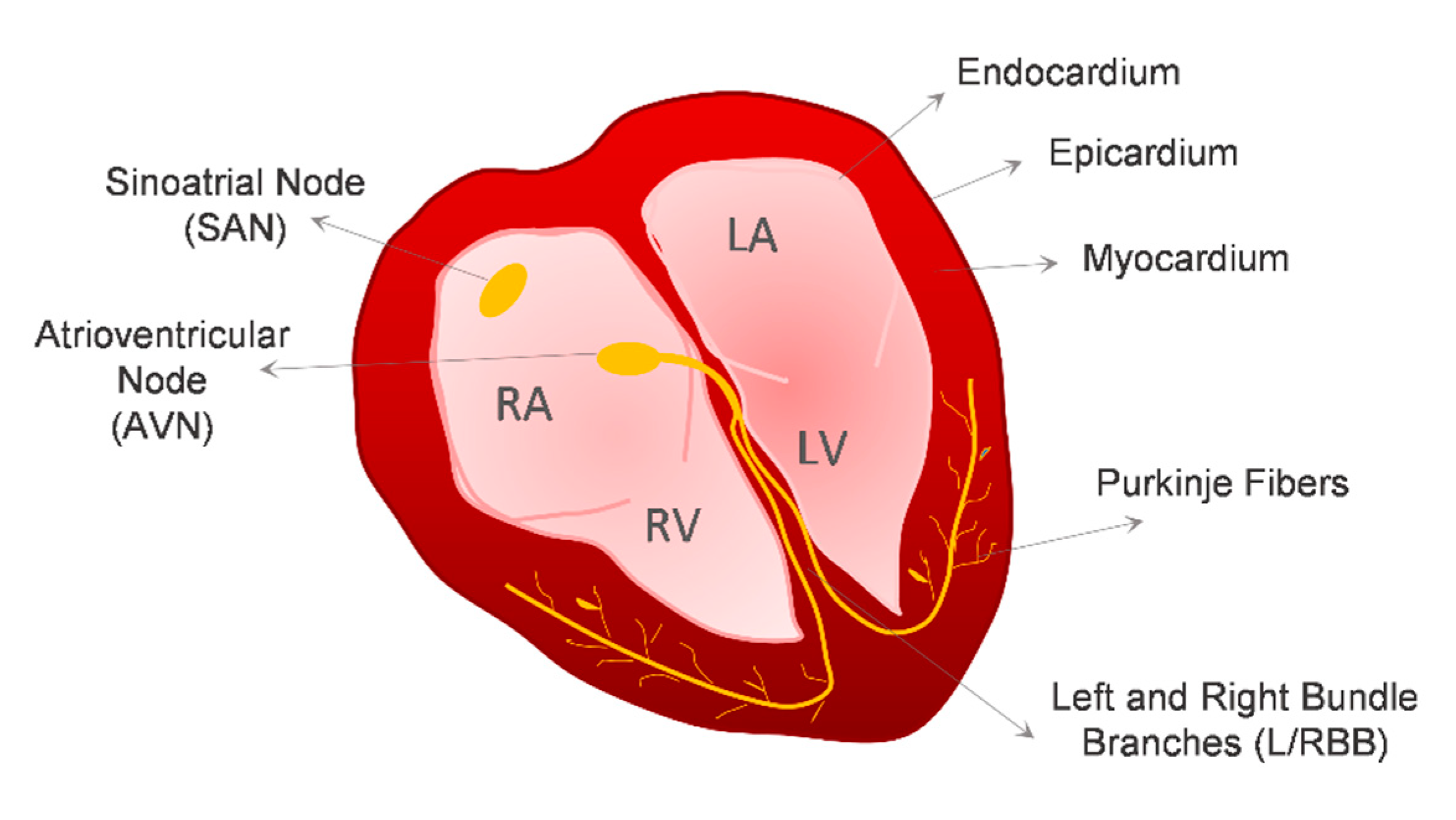
2. Cardiovascular Lineages Specification from hPSCs—Lessons from In Vivo Heart Development
2.1. Cardiomyocytes
2.2. Vascular Cardiac Cells


2.3. Cardiac Fibroblasts (CFs)
2.4. Epicardial Cells
3. Impact of 3D Environment on Cardiomyocyte Differentiation of hPSCs
|
Reference |
Pre-Differentiation |
Differentiation |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Platform |
Time |
Media |
Aggregate Ø at D0 |
Platform |
Media |
Molecules |
Duration |
Efficiency |
|||||
|
Halloin et al., 2019 |
Stirred Bioreactor |
2 Days |
E8 |
±125 µm |
Stirred Bioreactor (500 mL) |
CDM3 *1 |
CHIR |
D0–D1 |
5 µM |
10 Days |
±1 × 106 CMs/mL |
||
|
IWP2 |
D1–D3 |
5 µM |
93 ± 5% CMs |
||||||||||
|
Chen et al., 2015 |
Spinner Flask |
2 Days |
StemPro hESC SFM + FGF2 |
200 ± 20 μm |
Spinner Flask (125 mL–1L) |
RPMI + B27-INS |
D0–D4 |
CHIR |
D0–D1 |
6/12 µM *2 |
16 Days |
±1 × 106 CMs/mL (1L) ±2 × 106 CMs/mL (500 mL) |
|
|
RPMI + B27 |
D4–D16 |
IWP4 |
D2–D4 |
5 µM |
>90% CMs |
||||||||
|
Fonoudi et al., 2015 |
Stirred Bioreactor |
5 Days |
DMEM/F12+ FGF2 |
175 ± 25 µm |
Stirred Bioreactor (100 mL) |
RPMI + B27 |
D0–D15 |
CHIR |
D0–D1 |
12 µM |
15 Days |
0.8 × 106 CMs/mL |
|
|
SB + Pur + IWP2 |
D2–D4 |
5 µM each |
>80% CMs |
||||||||||
|
Branco et al., 2019 |
AggrewellTM800 |
3 Days |
mTeSR1 |
±300 µm |
AggrewellTM800 |
D0–D7 |
RPMI + B27-INS |
D0–D7 |
CHIR |
D0–D1 |
11 µM |
12 Days |
±20 × 106 CMs/plate |
|
ULA 6-well plate |
D0–D12 |
RPMI + B27 |
D7–D12 |
IWP4 |
D3–D5 |
5 µM |
>85% CMs |
||||||
|
Burridge et al., 2011 |
- |
*6 |
96-V ULA plate |
D0–D4 |
RPMI |
D0–D10 |
*5 |
10 Days |
±0.4 × 106 CMs/plate |
||||
|
96-U ULA plate |
D4–D10 |
>80% CMs |
|||||||||||
|
Dahlmann et al., 2013 |
Agarose Microwell plate |
1 Day |
FCM *3 |
400–500 µm *4 (±220 µm (D-3)) |
ULA 6-well plate |
RPMI + B27-INS |
D0–D7 |
CHIR |
D0–D1 |
8 µM |
10 Days |
*6 |
|
|
ULA 6-well plate |
3 Days |
RPMI + B27 |
D7–D10 |
IWR1 |
D3–D5 |
4 µM |
Up to 65% CMs |
||||||
*1 RPMI 1640 (+2 mM Glutamine) + 495 µg/mL Recombinant Human Albumin + 213 µg/mL Ascorbic Acid. *2 Depending on the cell line. *3 DMEM/F12 + GlutaMAX + 20% (v/v) Knockout serum replacement + 1% (v/v) non-essential amino acids + 0.1 mM β mercaptoethanol + 10 ng/mL FGF-2. *4 Determined by bright field image analysis. *5 D0-D2: BMP4 (25 ng/mL); FGF2 (5 ng/mL); PVA (4 mg/mL); h-Insulin (10 µg/mL). D2-D4: HAS (5 mg/mL); 280 µM L-ascorbic acid. D4-foward: h-Insulin (10 µg/mL). *6 Not specified*.
4. Engineering 3D Cardiac Microtissues to Better Mimic the Human Heart Environment
5. In Vitro Applications of hPSC-Derived 3D Cardiac Microtissues (MTs)

This entry is adapted from the peer-reviewed paper 10.3390/bioengineering7030092
References
- Camelliti, P.; Borg, T.K.; Kohl, P. Structural and functional characterisation of cardiac fibroblasts. Cardiovasc. Res. 2005, 65, 40–51.
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’Antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting Cardiac Cellular Composition. Circ. Res. 2016, 118, 400–409.
- Brutsaert, D.L. Cardiac Endothelial-Myocardial Signaling: Its Role in Cardiac Growth, Contractile Performance, and Rhythmicity. Physiol. Rev. 2003, 83, 59–115.
- World Health Organization. Available online: https://www.who.int/news-room/fact-sheets/detail/cardiovascular-diseases-(cvds) (accessed on 22 May 2020).
- Batho, C.A.P.; Mills, R.J.; Hudson, J.E. Metabolic Regulation of Human Pluripotent Stem Cell-Derived Cardiomyocyte Maturation. Curr. Cardiol. Rep. 2020, 22.
- Ahmed, R.E.; Anzai, T.; Chanthra, N.; Uosaki, H. A Brief Review of Current Maturation Methods for Human Induced Pluripotent Stem Cells-Derived Cardiomyocytes. Front. Cell Dev. Biol. 2020, 8, 1–9.
- Loh, K.M.M.; Chen, A.; Koh, P.W.W.; Deng, T.Z.Z.; Sinha, R.; Tsai, J.M.M.; Barkal, A.A.A.; Shen, K.Y.Y.; Jain, R.; Morganti, R.M.M.; et al. Mapping the Pairwise Choices Leading from Pluripotency to Human Bone, Heart, and Other Mesoderm Cell Types. Cell 2016, 166, 451–467.
- Loh, K.M.; Ang, L.T.; Zhang, J.; Kumar, V.; Ang, J.; Auyeong, J.Q.; Lee, K.L.; Choo, S.H.; Lim, C.Y.Y.; Nichane, M.; et al. Efficient endoderm induction from human pluripotent stem cells by logically directing signals controlling lineage bifurcations. Cell Stem Cell 2014, 14, 237–252.
- Rao, J.; Pfeiffer, M.J.; Frank, S.; Adachi, K.; Piccini, I.; Quaranta, R.; Araúzo-Bravo, M.; Schwarz, J.; Schade, D.; Leidel, S.; et al. Stepwise Clearance of Repressive Roadblocks Drives Cardiac Induction in Human ESCs. Cell Stem Cell 2016, 18, 341–353.
- Kattman, S.J.; Witty, A.D.; Gagliardi, M.; Dubois, N.C.; Niapour, M.; Hotta, A.; Ellis, J.; Keller, G. Stage-specific optimization of activin/nodal and BMP signaling promotes cardiac differentiation of mouse and human pluripotent stem cell lines. Cell Stem Cell 2011, 8, 228–240.
- Zhang, J.; Klos, M.; Wilson, G.F.; Herman, A.M.; Lian, X.; Raval, K.K.; Barron, M.R.; Hou, L.; Soerens, A.G.; Yu, J.; et al. Extracellular Matrix Promotes Highly Efficient Cardiac Differentiation of Human Pluripotent Stem Cells. Circ. Res. 2012, 111, 1125–1136.
- Hudson, J.; Titmarsh, D.; Hidalgo, A.; Wolvetang, E.; Cooper-White, J. Primitive Cardiac Cells from Human Embryonic Stem Cells. Stem Cells Dev. 2012, 21, 1513–1523.
- Uosaki, H.; Fukushima, H.; Takeuchi, A.; Matsuoka, S.; Nakatsuji, N.; Yamanaka, S.; Yamashita, J.K. Efficient and Scalable Purification of Cardiomyocytes from Human Embryonic and Induced Pluripotent Stem Cells by VCAM1 Surface Expression. PLoS ONE 2011, 6, e23657.
- Lian, X.; Hsiao, C.; Wilson, G.; Zhu, K.; Hazeltine, L.B.; Azarin, S.M.; Raval, K.K.; Zhang, J.; Kamp, T.J.; Palecek, S.P. Robust cardiomyocyte differentiation from human pluripotent stem cells via temporal modulation of canonical Wnt signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1848–E1857.
- Zhao, M.; Tang, Y.; Zhou, Y.; Zhang, J. Deciphering Role of Wnt Signalling in Cardiac Mesoderm and Cardiomyocyte Differentiation from Human iPSCs: Four-dimensional control of Wnt pathway for hiPSC-CMs differentiation. Sci. Rep. 2019, 9, 19389.
- Halloin, C.; Schwanke, K.; Löbel, W.; Franke, A.; Szepes, M.; Biswanath, S.; Wunderlich, S.; Merkert, S.; Weber, N.; Osten, F.; et al. Continuous WNT Control Enables Advanced hPSC Cardiac Processing and Prognostic Surface Marker Identification in Chemically Defined Suspension Culture. Stem Cell Rep. 2019, 13, 366–379.
- Kempf, H.; Olmer, R.; Haase, A.; Franke, A.; Bolesani, E.; Schwanke, K.; Robles-Diaz, D.; Coffee, M.; Göhring, G.; Dräger, G.; et al. Bulk cell density and Wnt/TGFbeta signalling regulate mesendodermal patterning of human pluripotent stem cells. Nat. Commun. 2016, 7, 13602.
- Burridge, P.W.; Thompson, S.; Millrod, M.A.; Weinberg, S.; Yuan, X.; Peters, A.; Mahairaki, V.; Koliatsos, V.E.; Tung, L.; Zambidis, E.T. A Universal System for Highly Efficient Cardiac Differentiation of Human Induced Pluripotent Stem Cells That Eliminates Interline Variability. PLoS ONE 2011, 6, e18293.
- Laco, F.; Woo, T.L.; Zhong, Q.; Szmyd, R.; Ting, S.; Khan, F.J.; Chai, C.L.L.; Reuveny, S.; Chen, A.; Oh, S. Unraveling the Inconsistencies of Cardiac Differentiation Efficiency Induced by the GSK3β Inhibitor CHIR99021 in Human Pluripotent Stem Cells. Stem Cell Rep. 2018, 10, 1851–1866.
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860.
- Lian, X.; Bao, X.; Zilberter, M.; Westman, M.; Fisahn, A.; Hsiao, C.; Hazeltine, L.B.; Dunn, K.K.; Kamp, T.J.; Palecek, S.P. Chemically defined, albumin-free human cardiomyocyte generation. Nat. Methods 2015, 12, 595–596.
- Tallquist, M.D.; Molkentin, J.D. Redefining the identity of cardiac fibroblasts. Nat. Rev. Cardiol. 2017, 14, 484–491.
- Moore-Morris, T.; Guimarães-Camboa, N.; Banerjee, I.; Zambon, A.C.; Kisseleva, T.; Velayoudon, A.; Stallcup, W.B.; Gu, Y.; Dalton, N.D.; Cedenilla, M.; et al. Resident fibroblast lineages mediate pressure overload-induced cardiac fibrosis. J. Clin. Investig. 2014, 124, 2921–2934.
- Zhang, J.; Tao, R.; Campbell, K.F.; Carvalho, J.L.; Ruiz, E.C.; Kim, G.C.; Schmuck, E.G.; Raval, A.N.; da Rocha, A.M.; Herron, T.J.; et al. Functional cardiac fibroblasts derived from human pluripotent stem cells via second heart field progenitors. Nat. Commun. 2019, 10.
- Witty, A.D.; Mihic, A.; Tam, R.Y.; Fisher, S.A.; Mikryukov, A.; Shoichet, M.S.; Li, R.K.; Kattman, S.J.; Keller, G. Generation of the epicardial lineage from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1026–1035.
- Bao, X.; Lian, X.; Hacker, T.A.; Schmuck, E.G.; Qian, T.; Bhute, V.J.; Han, T.; Shi, M.; Drowley, L.; Plowright, A.T.; et al. Long-term self-renewing human epicardial cells generated from pluripotent stem cells under defined xeno-free conditions. Nat. Biomed. Eng. 2017, 1, 1–12.
- Zhao, J.; Cao, H.; Tian, L.; Huo, W.; Zhai, K.; Wang, P.; Ji, G.; Ma, Y. Efficient Differentiation of TBX18+/WT1+ Epicardial-Like Cells from Human Pluripotent Stem Cells Using Small Molecular Compounds. Stem Cells Dev. 2017, 26, 528–540.
- Iyer, D.; Gambardella, L.; Bernard, W.G.; Serrano, F.; Mascetti, V.L.; Pedersen, R.A.; Talasila, A.; Sinha, S. Robust derivation of epicardium and its differentiated smooth muscle cell progeny from human pluripotent stem cells. Development 2015, 142, 1528–1541.
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct Compartments of the Proepicardial Organ Give Rise to Coronary Vascular Endothelial Cells. Dev. Cell 2012, 22, 639–650.
- Lupu, I.-E.; Redpath, A.N.; Smart, N. Spatiotemporal Analysis Reveals Overlap of Key Proepicardial Markers in the Developing Murine Heart. Stem Cell Rep. 2020, 14, 770–787.
- Cano, E.; Carmona, R.; Ruiz-Villalba, A.; Rojas, A.; Chau, Y.Y.; Wagner, K.D.; Wagner, N.; Hastie, N.D.; Muñoz-Chápuli, R.; Pérez-Pomares, J.M. Extracardiac septum transversum/proepicardial endothelial cells pattern embryonic coronary arterio-venous connections. Proc. Natl. Acad. Sci. USA 2016, 113, 656–661.
- Niebruegge, S.; Bauwens, C.L.; Peerani, R.; Thavandiran, N.; Masse, S.; Sevaptisidis, E.; Nanthakumar, K.; Woodhouse, K.; Husain, M.; Kumacheva, E.; et al. Generation of human embryonic stem cell-derived mesoderm and cardiac cells using size-specified aggregates in an oxygen-controlled bioreactor. Biotechnol. Bioeng. 2009, 102, 493–507.
- Burridge, P.W.; Anderson, D.; Priddle, H.; Barbadillo Muñoz, M.D.; Chamberlain, S.; Allegrucci, C.; Young, L.E.; Denning, C. Improved Human Embryonic Stem Cell Embryoid Body Homogeneity and Cardiomyocyte Differentiation from a Novel V-96 Plate Aggregation System Highlights Interline Variability. Stem Cells 2007, 25, 929–938.
- Bauwens, C.L.; Peerani, R.; Niebruegge, S.; Woodhouse, K.A.; Kumacheva, E.; Husain, M.; Zandstra, P.W. Control of Human Embryonic Stem Cell Colony and Aggregate Size Heterogeneity Influences Differentiation Trajectories. Stem Cells 2008, 26, 2300–2310.
- Mohr, J.C.; Zhang, J.; Azarin, S.M.; Soerens, A.G.; de Pablo, J.J.; Thomson, J.A.; Lyons, G.E.; Palecek, S.P.; Kamp, T.J. The microwell control of embryoid body size in order to regulate cardiac differentiation of human embryonic stem cells. Biomaterials 2010, 31, 1885–1893.
- Hwang, Y.S.; Bong, G.C.; Ortmann, D.; Hattori, N.; Moeller, H.C.; Khademhosseinia, A. Microwell-mediated control of embryoid body size regulates embryonic stem cell fate via differential expression of WNT5a and WNT11. Proc. Natl. Acad. Sci. USA 2009, 106, 16978–16983.
- Bauwens, C.L.; Song, H.; Thavandiran, N.; Ungrin, M.; Massé, S.; Nanthakumar, K.; Seguin, C.; Zandstra, P.W. Geometric control of cardiomyogenic induction in human pluripotent stem cells. Tissue Eng. 2011, 17, 1901–1909.
- Hsiao, C.; Tomai, M.; Glynn, J.; Palecek, S.P. Effects of 3-D microwell culture on initial fate specification in human embryonic stem cells. AIChE J. 2014, 60, 1225–1235.
- Azarin, S.M.; Lian, X.; Larson, E.A.; Popelka, H.M.; de Pablo, J.J.; Palecek, S.P. Modulation of Wnt/β-catenin signaling in human embryonic stem cells using a 3-D microwell array. Biomaterials 2012, 33, 2041–2049.
- Dahlmann, J.; Kensah, G.; Kempf, H.; Skvorc, D.; Gawol, A.; Elliott, D.A.; Dräger, G.; Zweigerdt, R.; Martin, U.; Gruh, I. The use of agarose microwells for scalable embryoid body formation and cardiac differentiation of human and murine pluripotent stem cells. Biomaterials 2013, 34, 2463–2471.
- Branco, M.A.; Cotovio, J.P.; Rodrigues, C.A.V.; Vaz, S.H.; Fernandes, T.G.; Moreira, L.M.; Cabral, J.M.S.; Diogo, M.M. Transcriptomic analysis of 3D Cardiac Differentiation of Human Induced Pluripotent Stem Cells Reveals Faster Cardiomyocyte Maturation Compared to 2D Culture. Sci. Rep. 2019, 9, 1–13.
- Chen, V.C.; Ye, J.; Shukla, P.; Hua, G.; Chen, D.; Lin, Z.; Liu, J.; Chai, J.; Gold, J.; Wu, J.; et al. Development of a scalable suspension culture for cardiac differentiation from human pluripotent stem cells. Stem Cell Res. 2015, 15, 365–375.
- Ting, S.; Chen, A.; Reuveny, S.; Oh, S. An intermittent rocking platform for integrated expansion and differentiation of human pluripotent stem cells to cardiomyocytes in suspended microcarrier cultures. Stem Cell Res. 2014, 13, 202–213.
- Fonoudi, H.; Ansari, H.; Abbasalizadeh, S.; Larijani, M.R.; Kiani, S.; Hashemizadeh, S.; Zarchi, A.S.; Bosman, A.; Blue, G.M.; Pahlavan, S.; et al. A Universal and Robust Integrated Platform for the Scalable Production of Human Cardiomyocytes From Pluripotent Stem Cells. Stem Cells Transl. Med. 2015, 4, 1482–1494.
- Kempf, H.; Olmer, R.; Kropp, C.; Rückert, M.; Jara-Avaca, M.; Robles-Diaz, D.; Franke, A.; Elliott, D.A.; Wojciechowski, D.; Fischer, M.; et al. Controlling Expansion and Cardiomyogenic Differentiation of Human Pluripotent Stem Cells in Scalable Suspension Culture. Stem Cell Rep. 2014, 3, 1132–1146.
- Hemmi, N.; Tohyama, S.; Nakajima, K.; Kanazawa, H.; Suzuki, T.; Hattori, F.; Seki, T.; Kishino, Y.; Hirano, A.; Okada, M.; et al. A Massive Suspension Culture System With Metabolic Purification for Human Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Transl. Med. 2014, 3, 1473–1483.
- Zhang, M.; Schulte, J.S.; Heinick, A.; Piccini, I.; Rao, J.; Quaranta, R.; Zeuschner, D.; Malan, D.; Kim, K.-P.; Röpke, A.; et al. Universal cardiac induction of human pluripotent stem cells in two and three-dimensional formats: Implications for in vitro maturation. Stem Cells 2015, 33, 1456–1469.
- Ma, Z.; Wang, J.; Loskill, P.; Huebsch, N.; Koo, S.; Svedlund, F.L.; Marks, N.C.; Hua, E.W.; Grigoropoulos, C.P.; Conklin, B.R.; et al. Self-organizing human cardiac microchambers mediated by geometric confinement. Nat. Commun. 2015, 6, 7413.
- Klesen, A.; Jakob, D.; Emig, R.; Kohl, P.; Ravens, U.; Peyronnet, R. Cardiac fibroblasts. Herzschrittmachertherapie + Elektrophysiologie 2018, 29, 62–69.
- Warmflash, A.; Sorre, B.; Etoc, F.; Siggia, E.D.; Brivanlou, A.H. A method to recapitulate early embryonic spatial patterning in human embryonic stem cells. Nat. Methods 2014, 11, 847–854.
- Thavandiran, N.; Hale, C.; Blit, P.; Sandberg, M.L.; McElvain, M.E.; Gagliardi, M.; Sun, B.; Witty, A.; Graham, G.; Mcintosh, M.; et al. Functional arrays of human pluripotent stem cell-derived cardiac microtissues. bioRxiv 2019, 566059.