Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Immunology
Several gene products play pivotal roles in the induction of inflammation and the progression of cancer. The Raf kinase inhibitory protein (RKIP) is a cytosolic protein that exerts pleiotropic activities in such conditions, and thus regulates oncogenesis and immune-mediated diseases through its deregulation.
- RKIP
- T cells
- cancer
- immunosuppression
- immunotherapy
- autoimmunity
1. Introduction
The Raf-1 kinase inhibitory protein (RKIP), also referred to as PEBP-1 or PBP, is a member of the phosphatidylethanolamine-binding protein (PEBP) family that was originally isolated from the bovine brain [1]. It is a small, cytosolic protein [2] with wide expression in the tissues of various mammalian species, including monkeys, rats, chickens, and humans [1,3,4,5,6,7]. RKIP, as a very dynamic protein with a flexible pocket loop, exists in a number of states to enhance its functional switch [8,9]. The RKIP molecule appears to have pleiotropic activities on multiple signaling pathways, thus critically affecting cellular processes associated with their activation [10,11,12].
Yeung et al., (2000) first identified RKIP as a physiological endogenous inhibitor of the Raf-1/MEK/ERK signaling pathway [13] via direct binding to all three MAP kinases. The binding of RKIP to both Raf-1 and MEK inhibits their phosphorylation and activation, thus leading to downstream suppression of the Raf-1-induced signaling and activity of AP-1-dependent transcription [13]. Furthermore, RKIP antagonizes the NF-kB signaling through its interaction with upstream kinases responsible for regulating the IkB protein while having a positive effect on the heterotrimeric G protein-coupled receptor (GPCR) and GSK signaling. RKIP also inhibits phosphorylation and activation of the transcriptional factor STAT3, suppresses the expression of NRF2-ARE containing genes, and enhances glycogen synthase kinase 3 beta (GSK3Beta)-dependent signaling [1,14,15,16,17,18,19]. Moreover, demonstrations have shown that the association of RKIP with the centrosomes and kinetochores plays a role in the regulation of the spindle checkpoint in mammalian cells [19].
Despite the well-established role of RKIP in cancer progression and aggressiveness, its role in tumor response to host immune-surveillance mechanisms and exogenous immunotherapy, as well as in the regulation of inflammatory responses, are less clear.
2. RKIP Structure
In humans, the RKIP mRNA molecule is 1434 base pairs (bp) long and shares no significant homology with other protein families [22]. It is transcribed from a gene that contains four exons of 10 kilo base pairs (kb) located in chromosome 12q24.23 [1,6,23,24]. This mRNA encodes a protein of 187 amino acids (aa) that shares sequences with bovine and rat RKIP [1,6,7]. The human RKIP protein consists of 23kDa and 186 aa [19]. Scientists have reported that RKIP crystalizes in two asymmetric molecules of 180 and 185 residues, although information regarding its functional oligomeric and dimeric states is unknown [19,22].
RKIP’s compact structure is composed of nine-stranded beta sheets and four alpha-helices folded into a pattern that enables it to be stable and gives it its unique properties, including its conserved ligand-binding pocket [19,25,26]. The pocket is composed of 16 aa residues and accommodates several different nucleotides, including phospholipids and non-lipid organic compounds, which contribute to the column-dependent purification of the molecule [19,27,28,29]. Amino acid residues 93–134 of RKIP are responsible for the formation of the Raf-1 binding domain, where the unphosphorylated and tri-phosphorylated forms of the Raf-1 protein bind to RKIP [19,25,26].
As mentioned above, RKIP exists in multiple states, which enhances its functional switch through its flexible pocket loop [8,9]. For instance, RKIP phosphorylation at Serine153 switches it from a Raf-1 binding state to a GRK2 binding state [8,18], thus exerting different functional roles in each of the implicated signaling pathways. Unlike the RKIP-GRK2 binding state (motif), the RKIP-Raf-1 interaction motif (state) has been crystallized in several mammalian models [8,9,22,30]. Studies have further reported that RKIP has a third state that interacts with phosphorylating kinases [31].
3. RKIP Functions on Cell Signaling
Below, we briefly describe the multiple functions of RKIP in cell signaling pathways with critical involvement in cell homeostasis, survival, and disease pathogenesis [32,33] (Figure 1).
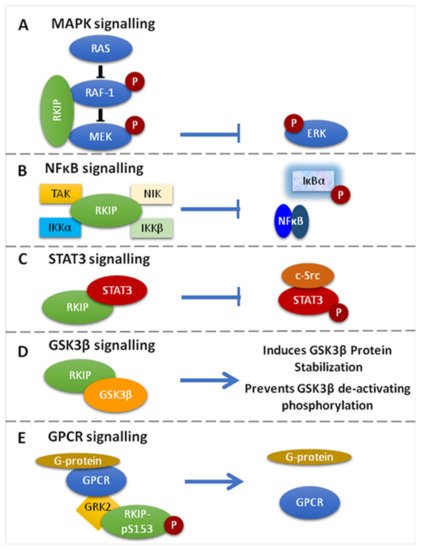
Figure 1. Signaling Pathways targeted by RKIP. (A). RKIP inhibits MAPK signaling via direct association with Raf-1 or MEK. The association prevents the phosphorylation and activation of the interacting kinases and therefore the downstream activation of ERK. (B). RKIP inhibits NF-κΒ signaling through interaction with the upstream kinases NIK, TAK, ΙΚΚα and IKKβ, resulting in the inhibition of IκΒα phosphorylation and the accumulation of NF-κΒ in the cytoplasm. (C). RKIP associates with STAT3 and inhibits its activation by preventing c-Src-mediated STAT3 phosphorylation. (D). RKIP interacts with GSK3β and promotes protein stabilization, while it inhibits its deactivating phosphorylation. (E). RKIP phosphorylated at Ser153 (RKIPpSer153) dissociates RAF-1 and binds GRK2, resulting in the de-repression of GPCR activation. Different coloured schemes in each panel represent different gene products.
3.1. RKIP-Mediated Inhibition of the Raf-1/MEK/ERK Pathway
The Ras/Raf-1/MEK/ERK pathway conveys mitogenic and differentiation signals to the nucleus [13,34]. It is organized in a complex that is nucleated by Ras proteins [35] that are activated by growth factors that bind the Raf-1 kinase with high affinity. These induce Raf-1 recruitment to the cell membrane and its activation via phosphorylation. Raf-1 activation, in turn, leads to the activation of MEK, which results in the phosphorylation and activation of ERK1/2 [13,36,37]. Activated ERKs migrate to the nucleus to regulate gene expression by phosphorylating transcription factors [13,38]. Yeast studies have revealed that the JIP-1 and Ksr proteins are important scaffolding molecules used to assemble components of the MAPK pathway [13,39]. Ksr, a protein kinase, functions by binding to Raf-1, MEK, and ERK, while JIP-1 serves as a scaffolding protein for the stress-activated MAPKs/JNKs [13,40,41,42,43,44,45,46,47]. The above studies helped in the identification of RKIP as an endogenous inhibitor of the MAPK pathway via physical association with different kinases of the cascade [13,48].
RKIP can specifically inhibit the Raf-1/MEK/ERK cascade by two mechanisms: (1) by binding to the N-region of the Raf-1 kinase domain, thus inhibiting its phosphorylation and activation, and (2) by dissociating the Raf-1/MEK complex, hence inhibiting the phosphorylation and activation of MEK [1,13,14,49,50] (Figure 1A). Interestingly, it was further shown that MEK and Raf-1 bind to the same sites on RKIP, even though MEK and RKIP are associated with different domains on Raf-1. Furthermore, it was shown that Raf-1 and RKIP bind to different sites on MEK. In order for the Raf-1/MEK/ERK pathway to be suppressed by RKIP, the MEK binding sites and the Raf-1 binding sites on RKIP must be deleted. This suggests that whether RKIP binds to Raf-1 or MEK, both are ample for their inhibition [13]. Given that the MAPK signaling promotes cell migration and RKIP inhibits MAPK signaling, the restoration of RKIP expression in human hepatoma (HHC) and melanoma cells repressed tumor cell migration and motility, as well as the cellular invasive potential [51,52,53,54].
3.2. Inhibition of the NF-kB Pathway
Active NF-kB acts as a transcriptional regulator of several genes that play a role in immunity, inflammation, cell proliferation, cell migration, and apoptosis [51,55]. RKIP is an inhibitor of NF-κB signaling by interacting with and inhibiting upstream activating kinases, such as the transforming growth factor B-activated kinase-1 (TAK-1), the NF-κB-inducing kinase (NIK), and the IκB kinaseσ (IKKα and IKKβ) [14] (Figure 1B). The translational impact of NF-κB inhibition by RKIP in cancer conveys with cancer cell sensitization to apoptotic signals mediated by chemotherapy, or endogenous immune-related cytotoxicity or exogenous immunotherapy, inhibition of EMT and cell metastatic potential, as well as reduced cell survival [51,56]. Researchers have also proposed that since RKIP interacts with members of the NF-κB signaling pathway, it could also serve as a scaffold protein that plays a role in assembling a multicomponent protein complex [51].
3.3. RKIP-Mediated Inhibition of GRK2
RKIP phosphorylation at Ser153 (pSer153RKIP) increases the survival and invasion of cancerous cells by inhibiting the G-protein-coupled receptor kinase 2 (GRK-2), which negatively regulates G-protein-coupled receptor (GPCR)-mediated signaling [1]. Briefly, under normal conditions, GRK-2 binds to GPCR and blocks its activation and intracellular signaling. When RKIP is unphosphorylated, it associates with Raf-1 and suppresses MAPK signaling. RKIP is phosphorylated at Serine 153 (S153) by GPCR-induced PKC zeta [57]. In turn, this phosphorylation dissociates RKIP from Raf-1, thus de-repressing RKIP-mediated MAPK inhibition. Subsequently, the phosphorylated RKIP binds to GRK2 and desensitizes GPCRs, leading to persistent phosphorylation of Raf-1 and activation of GPCR [18,57,58,59] (Figure 1E).
High levels of pSer153 RKIP are induced by IL-6 and H. pylori infection in colon and gastric cancers, respectively, and are associated with a poor prognosis in stage II colon cancer patients and little to no response to therapy for patients with multiple myeloma [57,60,61]. In gastric cancer models, H. pylori-mediated phosphorylation of RKIP at S153 promotes its nuclear accumulation in cancer cells, while it targets unphosphorylated RKIP for proteasome degradation [62,63]. Unlike the unphosphorylated RKIP form, the pRKIP acts by competitively inhibiting survival signals and promoting apoptosis in cancer cells [64,65,66].
3.4. RKIP-Mediated Inhibition of STAT3 Activation
STAT3 is a member of the signal transducer and activator of transcription (STAT) family, which is found in the cytoplasm, and upon being activated by phosphorylation, it translocates in the nucleus acting as a transcription factor for genes involved in the process of apoptosis, cell growth, survival, and differentiation [1,67]. Although the exact mechanism is unknown, studies in colon cancer models have shown that the overexpression of RKIP inhibits IL-6-, Jak-, or Src kinase-mediated phosphorylation of STAT3 known to be necessary for its activation [1,17] (Figure 1C).
3.5. Regulation of GSK3β Signaling
RKIP has been reported to regulate glycogen synthase kinase 3 (GSK3β) levels by (1) direct binding to the GSK3β protein that results in better protein stabilization and (2) by preventing GSK3β inhibitory phosphorylation [51] (Figure 1D). Depletion of RKIP induces high levels of oxidative stress response that leads to p38 MAPK activation, which ultimately inhibits GSK3β by phosphorylating its T390 residue, an inhibitory residue [51,68,69]. Furthermore, when RKIP is depleted, downstream GSK3β targets are activated, leading to cyclin D1 stabilization, which is what induces cell cycle progression and expression of β-catenin, SNAIL and SLUG. These three are also responsible for promoting the invasion and EMT [51,69]. In addition, in HEK-293 cells where RKIP was depleted, cell migration was favored by inducing p38-mediated phosphorylation of GSK3β, whereas its degradation stabilized cell migration regulatory molecules, such as β-catenin [51,69].
3.6. Regulation of the Spindle Checkpoint by RKIP
RKIP also regulates the spindle checkpoint and thus plays a role in the control of the cell cycle and the stability of the genome by associating with the centrosomes and kinetochores in mammalian cells [51,70,71]. RKIP-depleted cells rapidly move to the anaphase displaying a defective spindle checkpoint. Its depletion led to the reduced localization and kinase activity of Aurora B, a kinase that helps with chromosomal alignment as well as spindle checkpoints and cell division [51,70,72]. Furthermore, RKIP deficient cells tend to display decreased localization of Aurora B to kinetochores, which leads to inhibition of the activity of its kinase because of the hyperactivation of the MAPK pathway. This process sometimes leads to chromosomal abnormalities [51,70]. Using comparative genomic hybridization and allelotyping, researchers found that colorectal tumors lacking or weakly expressing RKIP display chromosomal losses and are genomically unstable, unlike cancers expressing it [51,73]. In addition, cells with depleted RKIP exert a shorter transition time from their nuclear envelope breakdown to anaphase, which, together with Aurora B and G2/M downregulation, gives cells a highly proliferative quality due to a faster rate of completion of the cell cycle phases [51,69]. This data further proposes that RKIP does in fact have an influence on cell proliferation and its overexpression reduces cell growth compared to RKIP depleted cells [51]. The depletion of RKIP indirectly influences the acceleration of cellular proliferation and growth by means of modulating the expression of genes that are involved in DNA replication, transition through G1/S phase, G2/M checkpoints, and genomic stability [51,69].
3.7. Clinical Significance of pRKIP in Various Cancers
3.7.1. pRKIP Expression Correlates with Good Prognosis
Huerta-Yepez et al., (2011) studied the expression of pRKIP and RKIP in NSCLC patients in order to determine the ability of these proteins to predict prognosis [65]. Using Western blot analysis, researchers studied three different lung cancer cell lines to see the levels of pRKIP and RKIP, which they found to be different for all the 3 cell types [65]. Although RKIP expression was constant in nonmetastatic and invasive and metastatic cases, pRKIP expression was decreased in invasive cancers compared to non-malignant cancers [65]. Furthermore, the significance of RKIP and pRKIP levels as a predictor of prognosis was tested via the Cox model analysis and was first found to have no significant effect on prognosis. A second trial was run using dichotomized RKIP and pRKIP expression and it was found that a higher expression of pRKIP did in fact lead to a greater survival of patients with NSCLC compared to those with lower expression of pRKIP [65]. More specifically, researchers found that patients that had higher levels of pRKIP were mostly older than 65 and that in patients younger than 65, high levels of pRKIP were not necessarily an indicator of survival. In addition, high levels of pRKIP served as a marker of good prognosis in early-stage NSCLC but not in the later stages of the disease [65].
Studies have also shown that a loss or reduction in phosphorylated RKIP expression in patients with breast cancer is interrelated with poor disease-free survival [64,65]. A study done by Al-Mulla et al., determined that when there was a reduction in RKIP expression or a loss of expression, patients with breast cancer often times were prone to larger sized tumors with substantial necrosis and a higher tumor grade [51]. Furthermore, by looking at data from 115 women with breast cancer that were previously studied, they found that in women with higher RKIP gene expression, the cancer cells tended to be non-metastatic while those with lower gene expression had cells that became metastatic [51]. Patients with low RKIP expression also exhibited significantly lower disease-free survival compared to patients with tumors that had high RKIP mRNA levels. These data suggest that higher levels of pRKIP, which lead to higher expression of RKIP, correlate with a good prognosis. However, limited and contradictory data are available when it comes to the clinical implications of pRKIP in tumors. A study done by Li et al., (2016) was the first to show that pSer153RKIP is a favorable prognostic factor for patients with nasopharyngeal carcinoma (NPC) who received radiation [64], while in patients with early-stage lung cancer, normal expression of phosphorylated RKIP was an indicator of more favorable survival [51,64]. On the other hand, RKIP is phosphorylated in other tumors, including multiple myeloma and stage II colon cancer, positively contributing to cell survival and drug resistance, which is mainly done through the transcription of downstream anti-apoptotic genes [60,64,66].
3.7.2. pRKIP Expression Correlates with Poor Prognosis
Cross-Knorr et al., (2013) performed a study demonstrating the effects of IL-6-mediated activation of STAT3 on the phosphorylation of RKIP [60]. They performed this study on colon cancer cells of humans. Increased IL-6 stimulation is common in various cell lines and tumors and is linked to cancer metastasis and cancer cell survival as a result of STAT3 phosphorylation [60,74,75,76,77,78,79,80]. Specifically, in colon cancer there is often an increase in the soluble receptor of IL-6 rather than the membrane receptor, leading to an increase in the activation of STAT and hence the activation of pro-survival proteins [60,81,82]. The activation of STAT3 by IL-6 in colon cancer was studied by examining HCT116 cells after IL-6 treatment, and specifically looking at STAT3 and pRKIP levels [60]. Furthermore, after questioning the effects of STAT3 overexpression on transcription and pRKIP, Cross-Knorr et al., performed Western blot analyses and found that when transfected with STAT3, the levels of expression of phosphorylated RKIP did in fact increase [60]. This increase in expression has led to a poor prognosis in patients with stage II colon cancer. A Kaplan–Meier analysis was done on a group of patients and indicated that elevated pRKIP was correlated with a decrease in the survival of patients and that patients with lower levels of pRKIP had lower levels of lymphovascular invasion than those with higher levels of pRKIP [60].
This study had different results to the study done by Huerta-Yepez et al., who found that in patients with non-small cell lung cancer, those who have normal pRKIP levels are more likely to have a better prognosis [83]. It is hypothesized that these two studies may have had different results because the experiments were run using different types of tissues or because when pRKIP is phosphorylated, a number of distinct pathways can be activated, which would result in worse or better prognosis in patients depending on which pathways were active. In conclusion, patients with stage II colon cancer exhibiting high levels of pRKIP had poor prognosis and shorter recurrence-free survival [60].
4. Regulation of RKIP Expression
Below, we describe the different ways that RKIP is regulated and the different regulators responsible for its regulation (Figure 2).
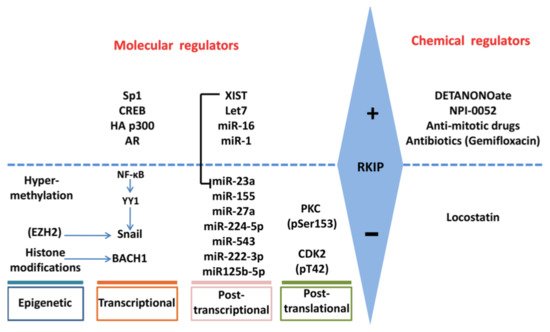
Figure 2. Regulation of RKIP expression. RKIP expression is regulated positively or negatively by multiple gene products and processes at epigenetic, transcriptional, post-transcriptional and post-translational levels. The involvement of each molecular regulator is cancer-type dependent. RKIP levels may also be modified indirectly by chemical molecules that may target the direct RKIP regulators.
4.1. SNAIL
The most well-known transcriptional regulator of RKIP is the epithelial-to-mesenchymal transition (EMT) protein SNAIL. Researchers have demonstrated a negative correlation between SNAIL and RKIP expression in prostate cancer and identified SNAIL as a direct transcriptional repressor. It was proven that SNAIL expression can be induced by NFκB and the transcription factor Yin Yang 1 (YY1), leading to RKIP downregulation [62,84,85]. Furthermore, in melanoma cell lines, a PDZ-domain (the postsynaptic density protein (PSD-95), melanoma differentiation associated gene 9 (MDA-9), discs-large tumor suppressor (Dlg), and tight junction protein-1 (ZO-1)) contains a scaffold protein that can silence RKIP transcription by activating SNAIL expression through ERK1/2 and NFκB signaling due to its role in promoting melanoma progression [54,62].
As for the chemical induction of RKIP, the organic molecule nitric oxide was found to be able to induce RKIP expression [62,86,87]. A nitric oxide donor, DETANONOate, inhibits NFκB signaling and lifts SNAIL-mediated transcriptional suppression on the RKIP promoter [62,86]. In some cases, chemotherapy, immunotherapy, and radiotherapy can also induce RKIP expression.
Snail has been shown to strongly repress E-cadherin transcription. Due to the coexpression between RKIP and E-cadherin, the hypothesis was that Snail may also act similarly towards RKIP [84,88,89]. Suppression of RKIP expression is inversely correlated with the expression levels of Snail; thus, when Snail expression was knocked down by specific siRNA, RKIP expression increased, suggesting that Snail is a repressor of RKIP [84]. Snail represses RKIP by binding to the E-box cis-elements in the RKIP promoter and recruiting mSin3A histone deacetylases and transcriptional repressor complexes [84,90,91]. When Snail is present, the enhancer of Zeste homolog 2 (EZH2) inhibits RKIP expression at the transcriptional level.
4.2. BACH1
BACH1 expression is negatively correlated with the expression of RKIP in breast cancer, suggesting that it may be its key negative regulator [62]. Similar to Snail, BACH1 positively correlates with the expression of EMT-associated genes, implying that it plays a role in EMT [62]. In addition, both molecules negatively regulate their own promoters, reducing their expression in a negative feedback loop manner. In addition, both are downstream targets of RKIP but also its negative regulators [62,92].
4.3. SP1, CREB, p300, AR
The region spanning from −56 to +261 nucleotides in the promoter region of RKIP is needed for its full activity. In this region, cis-acting elements that respond to Sp1, CREB, and histone acetylase p300 are needed for the maintenance of its promoter activity [57,93]. For example, studies have shown that knocking down CREB, p300, or Sp1 or the mutation or deletion of these elements limited RKIP promoter activity, meaning that they positively regulate RKIP transcription [57,93]. Finally, in prostate cells, the binding of the androgen receptor to an androgen responsive element in the RKIP promoter has a positive regulatory effect on the regulation of its transcription [57,94].
4.4. c-MET
The transcription factor C-MET activates β-catenin and acts as a growth factor that promotes cell migration, whereas PAK1 is a protein kinase involved in cell motility. Both molecules were found to be upregulated in RKIP-silenced HEK-293 cells [51,95,96], supporting the idea that RKIP loss influences the cell proteome and transcriptome to favor migration.
4.5. MMPs
It has been hypothesized that RKIP’s suppressive role in metastasis and cell migration is due to its ability to downregulate specific matrix metalloproteinases (MMPs) expression, such as MMP-1 and MMP-2 [51,97]. High expression of MMP-1 and MMP-2 along with a high invasive cellular property as a result of RKIP silencing led to the conclusion that RKIP controls the invasion of cancer cells by negatively regulating NF-kB, which in turn controls the expression of MMP and, therefore, cellular invasion [51,97].
4.6. EZH2
It is known that the downregulation of EZH2 inhibits cancer cell growth, proliferation, and invasion [98,99]. A decreased EZH2 expression can also increase the expression of RKIP in cancer cells [98]. An experiment was done to test whether decreasing cellular proliferation, growth, or invasion via silencing EZH2 could be reversed if RKIP expression was silenced. The inhibition of RKIP had no effect on cellular growth or proliferation, but it did effectively reverse the decrease in invasiveness that was caused by the loss of EZH2 [98], suggesting that the EZH2-mediated inhibition of RKIP is part of the molecular mechanism by which EZH2 promotes cellular invasion and metastasis in prostate and breast malignancies.
4.7. Methylation
The quantification of RKIP transcript levels in different cancer cell lines suggested that its downregulation is due to changes in the stability of its mRNA or the initiation of transcription [84]. Another study was performed to determine whether RKIP is repressed by methylation in metastatic prostate cell lines by examining the effects of trichostatin A (TSA, a histone deacetylase (HDAC) inhibitor) on RKIP expression. The authors reported that TSA significantly increased RKIP expression in the human prostate carcinoma cell line, DU145, but treatment with 3 microM 5-Aza-2dC, a demethylation agent, had no effect on RKIP expression [84,100]. The cellular treatment with 5-Aza-2dC showed that hyper-methylation is not the cause of RKIP downregulation. In addition, the fact that RKIP expression can be induced by TSA in cancer cells implies that it may be actively repressed in them [84]. Several studies on the promoter methylation have shown that the methylation status of the RKIP promoter is correlated with low RKIP expression levels in advanced stages of several tumors, including gastric adenocarcinomas, esophageal squamous cell carcinomas, and colorectal and breast cancers [62,73,101,102,103,104,105,106]. In esophageal and gastric cancers, the RKIP promoter was significantly hypermethylated in poorly differentiated tumors and lymph node metastases and this hypermethylation was associated with worse overall survival [62,102,104]. Furthermore, in the prostate cancer cell line DU145, TSA caused an increase in RKIP levels [62,84], which raises the question of whether histone deacetylation plays a role in RKIP silencing or not. One study showed that treatment of the triple-negative breast cancer (TNBC) cell line SUM159 with histone deacetylase inhibitors induced RKIP expression [107], but this was not verified in a different study [92]. Further research on this noted that BACH1 was induced by treatment with an HDAC inhibitor [62,92]. The authors also confirmed that EZH2 interacts with BACH1 in the TNBC cells to inhibit RKIP transcription, which can also be seen in breast and prostate cancer cells by interaction with Snail [62,98].
4.8. miRNAs
As seen in previous studies, RKIP’s expression is inversely correlated with miR-23a in prostate cancer and other malignancies [108,109,110]. To confirm the involvement of miR-23a in the regulation of RKIP in prostate cancer, Du et al., (2017b) computationally predicted that miR-23a binds directly to the 3′UTR of RKIP [108]. This prediction was verified using luciferase reporter assays. Co-transfection with luciferase reporter plasmids and miR-23a mimics showed that the luciferase activity of RKIP was significantly inhibited [108]. The knockdown of miR-23a also led to a significant increase in the expression of RKIP, whereas over-expression of miR-23a caused a significant reduction in RKIP protein expression [108]. Overall, the above results indicate that miR-23a negatively regulates RKIP in prostate cancer cells. Furthermore, RKIP gene expression can be inhibited by certain microRNAs [111], but the overexpression of let-7, miR-1, and miR-16 can enhance RKIP protein translation. On the other hand, the overexpression of miR-155 can destabilize RKIP and reduces its expression [90,91,112,113].
RKIP is reduced in metastasis and has been especially studied in breast cancer cells [114,115]. Several miRNAs, including miR-224, miR-27a, miR-23a, and miR-543 have been shown to target and inhibit the RKIP transcript [102,108,110,114,116,117]. Of these, only miR-224 seems to inhibit RKIP expression in breast cancer [111,114]. After looking at the relationship of RKIP expression and 2238 miRNAs, researchers classified three miRNAs as putative RKIP expression-regulating molecules: miR-224-5p, miR-222-3p and miR-125b-5p [110,111,114]. These were selected based on their correlative expression with RKIP in breast cancer cell lines [84,114]. To establish a regulating effect of the miRNAs on RKIP, three conditions need to be applied. They are: (i) negatively correlated with its expression levels, (ii) physically associated with the RKIP mRNA molecule, and (iii) the existence of miRNA recognition elements (MREs) for the identified miRNA in the 3′UTR and amino acid coding sequence of the RKIP gene is required [110,114]. Experiments showed that miR-224-5p functions as an RKIP repressor in breast cancer cells [111,114,118].
The miR-224 can negatively regulate RKIP, contributing to increased cell proliferation and invasion in gastric and breast tumors and in hepatocellular carcinoma [57,117,119,120]. RKIP can also be targeted by miR-27a, the upregulation of which contributes to chemoresistance in lung adenocarcinoma [57,117].
4.9. PKC
The protein kinase C (PKC) phosphorylates RKIP, activating its downstream signaling pathways [121]. PKC signal transduction participates in the process of T cell apoptosis and proliferation. It is the key enzyme during the cells inner signal transduction, which can regulate the expression of IL-2, IL-6, GM-CSF, and IL-1B by means of activating the backward position of NF-kB and AP-1 [122]. PKC also plays an important role in the immune inflammation reaction, cell apoptosis and proliferation, and self-immunity disease. PKC also plays a role in the mechanisms by which asthma works [122,123]. PKC phosphorylates RKIP at serine 153, dissociating it from Raf-1 [124].
4.10. XIST
Du et al., (2017b) investigated whether x-inactive specific transcript (XIST) acts as a competing endogenous RNA (ceRNA) in RKIP regulation, using immunoblotting assays to determine its expression post transfection with XIST (or si-XIST) and miR-23a mimics (or inhibitors, respectively) [108]. The results revealed that XIST overexpression substantially promoted RKIP expression, whereas miR-23a repudiated it [108]. While this was the case for overexpression of XIST, researchers found that the knockdown of XIST led to a reduction in RKIP that could essentially be returned by the inhibition of miR-23a [108]. All in all, these findings suggest that lncRNA XIST regulates RKIP expression in a ceRNA manner and miR-23a plays a main role in XIST-mediated regulatory pathways.
XIST has been known to be responsible for suppressing cellular proliferation and metastasis in prostate cancer cell lines and for negatively regulating miR-23a expression [108]. While investigating the correlation between XIST and miR-23a, miR-23a was identified as a direct target of XIST and its over-expression could overturn the XIST-induced RKIP up-regulation [108]. This suggests that XIST positively regulates RKIP expression through miR-23a binding.
5. Effects of RKIP Expression Levels in Various Cancers
In humans and other mammals, NF-kB binds to the promoter and enhancer sequences in various cells. It plays a known role in immune response, inflammatory response, and cell growth regulation through its ability to regulate adhesion molecules, immune receptors, and chemotactic and growth factors [125,126]. This study also showed that NF-kB enhances the host defense function of macrophages and neutrophil granulocytes and has a function in antigen presentation in dendritic cells and T cell activation [125]. Therefore, the degradation of NF-kB or the knockdown of NF-kB-dependent genes can lead to immune dysfunction in the host [127], and the overexpression of RKIP can lead to a decreased level of NF-kB-mediated responses [125]. In healthy patients, the immune system maintains a stable state with T-lymphocytes and Treg cells. It has been useful to understand that the gene products of mice known as cluster of differentiation (CD) can recognize the antigenic determinant on the surface of these T lymphocytes [125]. CD3 is the surface marker for mature T cells, CD4 is the marker of T helper cells, and CD8 is the marker of cytotoxic T cells. A decline in CD3 cells and an abnormal CD4/CD8 ratio indicate impaired cellular immunity [125]. Wei et al., (2015) found that CD3+ and CD4+ cell counts and CD4+/CD8+ ratios in gastric cardia adenocarcinoma cases were lower than in the control [125]. They also noted that CD8+ and Treg cell counts of gastric cardia adenocarcinoma were higher than in the control. These variations were all related to the levels of RKIP expression. Overall, it was hypothesized that the downregulation or deletion of RKIP has an effect on the NF-kB-mediated positive feedback mechanism and leads to the activation of upstream regulators [125,128]. The deletion of these genes, in turn, causes the inhibition of NF-kB activity and a steep decline in cellular immunity. In conclusion, RKIP expression is able to closely associate itself with metastasis and progression of gastric cardia adenocarcinoma tumors because of the inhibition of NF-kB activity and cellular immunity, which enables tumor cells to evade immune surveillance.
RKIP plays a major role in the survival of cancer patients. Two hyperactivated pathways in cancers are the MAPK/ERK and NF-kB pathways, both being regulated negatively by RKIP. The ERK1/2 are well-known as downstream effectors of the MAPK pathway that phosphorylate and activate several transcription factors, including CREB, c-Myc, and NF-kB, all of which play a role in regulating cell proliferation, differentiation, and survival [129,130]. In addition to suppressing the activating phosphorylation of ERK, RKIP also inhibits cell proliferation and promotes cell death by ablating MAPK signaling [48,129,130,131,132,133,134]. More specifically, the loss of RKIP function correlates with the inactivation of aurora B kinase through the hyperactivation of the Raf/MEK/ERK signaling cascade [70,129]. Interestingly, bypassing this checkpoint can result in chromosomal abnormalities, and the extent of genomic instability is measured by chromosomal losses, which are inversely proportional to the expression levels of RKIP in colorectal cancer [73,129].
NF-kB is a pro-survival transcription factor that exerts its activity through effector proteins and RKIP has a direct influence on its activation by inhibiting upstream kinases including NIK, TAK1, and IKK [14,129]. The inactivation of NF-kB by RKIP is important because it is most notably a participant in the pro-survival and anti-apoptotic pathway called the NF-kB/Snail/YY1/RKIP/PTEN dysregulated loop [129,135]. The induction of RKIP overexpression is able to interrupt the anti-apoptotic natural properties of the loop [136,137], while Snail and YY1, NF-kB-regulated factors, promote anti-apoptotic and pro-metastatic genes and serve as negative regulators of RKIP expression [129,138]. Another important molecule that plays a role in the regulation of cell cycle and survival is the transcription factor STAT3. RKIP blocks IL-6-, Janus kinase 1/2 (JAK1/2)-, and Raf-mediated activation of STAT3 by c-Src and c-Src autophosphorylation [129,139]. Overexpression of RKIP, in turn, enhances apoptosis by suppressing STAT3 targets. From a clinical standpoint, high levels of nuclear pRKIP and STAT3 are correlated with poor prognosis in stage II colon cancer patients, suggesting that RKIP regulates STAT3-mediated cell survival [60,129].
Ying Yang 1 (YY1) promotes therapeutic resistance in solid and hematological malignancies [12,134,135,140,141,142]. YY1 is regulated by the inhibition of NF-kB by RKIP since it functions downstream of NF-kB. This, in turn, eliminates the effect that it has on the resistance of cancer cells [12,57,135,143]. YY1 inhibition induced by RKIP contributes to Snail suppression since YY1 is known to directly act as a Snail transcription activator [12,57,136,144]. Therefore, YY1 might function as a link between NF-kB and Snail activation, which affects the activity of downstream cell death pathways [12,85,135]. Because of this, RKIP’s ability towards the NF-kB/YY1/Snail circuit is thought to be the underlying mechanisms of RKIP-mediated inhibition of tumor chemoresistance and immune-resistance [12,57,135].
Below, we give a few examples of different cancers in which RKIP plays a role (Table 1).
Table 1. RKIP expression in various cancers and their functions.
| Expression of RKIP | Impact/Functions | References | |
|---|---|---|---|
| Adenocarcinomas | Decreased RKIP expression | RKIP increases progression, metastasis, and invasion leading to a poor prognosis | Wei et al., 2015; Wei et al., 2014 |
| Colon Cancer | Reduction of RKIP | Amplifies radio-resistance and chemoresistance | Zaravinos et al., 2018; Lee et al., 2016 |
| Prostate Cancer | RKIP is downregulated | Enhances metastasis in prostate cancer-associated cell lines | Beach et al., 2008 |
| Pancreatic Cancer | RKIP is induced | Prevents the invasive metastasis of pancreatic cancer cells | Kim and Kim, 2012 |
| Gliomas | Low expression of RKIP | Does not affect cell proliferation, and enhances cell migration | Martinho et al., 2012 |
| Renal Cell Carcinoma | High RKIP expression | Induces cell survival and progression-free survival | Papale et al., 2017 |
| Gastric Cancer | Low levels of RKIP expression | Negatively correlated with depth of tumor invasion | Wang et al., 2010 |
| Lung Cancer | Decreased levels in invasive cancers | Greater advantage in survival | Huerta-Yepez et al., 2011 |
| Leukemia | Loss of RKIP expression is common | RKIP inhibits proliferation of myeloid cells | Zebisch et al., 2019 |
| Multiple Myeloma | RKIP is overexpressed | Enhances tumor progression | Shvartsur et al., 2017 |
5.1. Adenocarcinomas
RKIP is widely distributed across different human tissues [12,23,53,115,125]. In patients with adenocarcinoma, the deletion or downregulation of the RKIP gene was predicted to result in a poor prognosis [125]. Scientists found that in gastric cardia adenocarcinoma, the expression of RKIP was substantially lower in precancerous tissues in patients with lymph node metastasis, which further implies that RKIP plays a role in the progression, metastasis, and invasion of gastric cardia adenocarcinoma [125,145].
5.2. Colon Cancer
A further study showed that the overexpression of Snail correlates with high expression of cancer stem cell (CSC) markers and increased chemoresistance in colon cancer cell lines [12]. When looking at the effects of Snail silencing and RKIP upregulation, it was seen that the Snail/RKIP loop is an essential component of CSC existence within the tumor, associated with regulation and tumor chemoresistance [12,146]. The reduction of RKIP led to amplified radio-resistance of non-small-cell lung cancer (NSCLC), which generally accelerates the expression of CSC markers and sustains properties of CSC via the expression of Snail [12,147,148]. This demonstrates that RKIP has a negative effect on radio-resistance regulation and chemoresistance because it affects the number and function of CSCs in the tumor.
5.3. Prostate Cancer
Initially, RKIP was found to play a role in cancer through a gene array analysis that was performed to determine the genes that regulate metastasis. Researchers discovered that the prostate cancer (PCa)-associated LNCaP cell line has a low metastatic rate and therefore expresses higher RKIP levels than the cells of the derivative cell line C4-2B, which contained a high metastatic rate [1,23]. When C4-2B PCa cells were transfected with RKIP, its expression was restored and led to a reduction in spontaneous lung metastasis, but not primary tumor growth, proving that RKIP functions as a metastasis suppressor gene [1,56,149]. In addition, the restoration of RKIP expression was shown to inhibit breast cancer metastasis in murine models [91,97]. The downregulation of RKIP induced a high viability and migration of cells but did not have an effect on angiogenesis and cellular proliferation [1,150]. RKIP acts as an inhibitor at the molecular level and its loss promotes metastasis through the inhibition of angiogenesis, local invasion, colonization, and intravasation [1,23,91]. It is possible that the inhibitory protein does this by modulating the extracellular matrix [1,151]. RKIP also acts as a phosphorylation target for pathways that involve MAP kinase (MAPK) and the beta-adrenergic receptor (B-AR) and regulates these pathways by binding as well as inhibiting Raf and G protein-coupled receptor kinase 2 (GRK2) [8,18].
It has been found that RKIP expression is downregulated in several tumors, including those of highly metastatic prostate, colon, and breast cancer, hepatocellular carcinoma, and skin melanomas [12,23,52,53,56,84,115,149,152,153]. Furthermore, the restoration of its expression inhibits prostate cancer metastasis [23,84,154]. In order to study the regulation of RKIP, researchers examined its expression in cancer cell lines with different metastatic capacity. They observed that in highly invasive, metastatic cancers, such as breast and prostate cancers, its expression is repressed, while in noninvasive cell lines RKIP expression is high [84]. Researchers also found that RKIP is correlated with the intracellular adhesion protein E-cadherin (E-cad), which is regulated by the Snail and Slug transcription factors [84,155].
Prostate cancer accounts for 15% of cancers diagnosed in males and for 13% of cancer-related deaths [108,156]. In early state prostate cancer, tumor growth is generally attributed to the presence of androgens in the body. Androgen deprivation therapy (ADT) is the main form of treatment used for androgen-dependent prostate cancer, but a large majority of the tumors tend to continue to grow after remission that lasts about 18–24 months and continue to do so in an androgen-independent manner [108,157,158]. The long non-coding RNAs (lncRNAs) are a relatively new form of non-encoding RNA transcripts (>200 nucleotides) [108,159] that participate in cellular development and differentiation, as well as in tumorigenesis. They can also regulate gene expression in multiple ways, thus affecting several processes, including chromatin structure, nuclear transport, cutting and splicing, transcriptional modification, epigenetic control, and RNA decay [108,160,161,162,163,164,165]. LncRNAs have also been observed in many cancers and studies indicate that they can act as tumor suppressors, oncogenes, or even both [108,166]. For instance, Zeng et al., (2017) reported that lncRNA AF113014 acts as a tumor suppressor in hepatocellular carcinoma cells by promoting Egr2 expression [167], and Li et al., (2017) showed that the lncRNA n340790 enhances cellular proliferation in thyroid cancer by means of targeting miR-1254 [168]. In a study by Du et al. (2017), it was confirmed that the downregulation of XIST in prostate cancer is a common molecular change [116]. The expression of XIST is negatively correlated with metastasis and its low expression is associated with poor prognosis in prostate cancer patients. Furthermore, the overexpression of XIST has the potential to inhibit proliferation, migration, and invasion in prostate cancer cells [108].
Scientists have found evidence that the expression of lncRNAs is strongly associated with the development of cancer. Studies have shown that the lncRNA XIST regulates different cancers but in prostate cancer its underlying mechanism is still unclear [108]. A downregulation of XIST in prostate cancer specimens and cell lines that leads to a low expression of XIST has been correlated with advanced tumor stage in patients with prostate cancer and a poor prognosis in those patients [108].
5.4. Pancreatic Cancer
A study done in 2012 aimed at assessing whether (-)-epigallocatechin 3-gallate (EGCG) regulates the expression of RKIP and invasive metastatic activity in AsPC-1 pancreatic adenocarcinoma cells via epigenetic modifications. Results showed that RKIP expression differed in human pancreatic cancer cell lines. Baseline levels of RKIP were investigated in the cell lines MIA PaCa-2, PANC-1, AsPC-1, and BxPC-3. The AsPC-1 cell line was specifically selected to find out whether RKIP expression was in fact induced by EGCG treatment since it had the lowest level of RKIP of all the chosen cell lines [169]. An MTT assay was used to examine the effect of EGCG on AsPC-1 viability and it was revealed that no toxicity was present up to 10 μM EGCG, which is less than the usual EGCG treatment which is 15 μM [169]. Treatment with 10 μM of EGCG for 24 h led to an increase in RKIP expression in AsPC-1 cells compared to control cells, and in order to confirm if this RKIP regulation was due to histone modification, researchers performed the same experiments along with cellular treatment with TSA [169]. They found that in the presence of TSA, RKIP expression was induced and that the effects were synergistic to the effects of EGCG, allowing them to conclude that in AsPC-1 cells treated with EGCG, RKIP induction is partly due to HDAC modifications [169]. Furthermore, to explain the mechanism in which EGCG inhibits invasion and metastasis, researchers studied the expression of metastasis-related genes, such as MMP-2 and -9, Snail and E-cadherin. Results revealed that compared to the mRNA and protein levels in the control cells, the levels in MMP-2, MMP-9, pERK, and Snail were downregulated in the EGCG-treated AsPC-1 cells [169]. E-cadherin was notably increased by EGCG treatment. Since RKIP is responsible for regulating NF-kB activation via the MEK/ERK signaling pathway, the next step was to investigate whether treatment with EGCG inhibits the ERK phosphorylation [169,170]. The results of this experiment showed that treatment of cells with EGCG suppressed ERK phosphorylation and increased RKIP expression. Essentially, the results proved that when RKIP expression is induced by EGCG, it in turn inhibits the phosphorylation of ERK and NF-κB activation while also decreasing Snail expression, which means that EGCG acts as an HDAC inhibitor and can prevent the invasive metastasis of human PC cells. In conclusion, the results of this study showed that EGCG induced RKIP upregulation through the inhibition of HDAC activity, which in turn increases histone H3 expression and inhibits Snail expression, NF-κB nuclear translocation, MMP-2 and MMP-9 activity, and Matrigel invasion in AsPC-1 cells [169]. The results also infer that EGCG regulates RKIP/ERK/NF-κB and/or RKIP/NF-κB/Snail, as well as inhibiting invasive metastasis in the AsPC-1 human pancreatic adenocarcinoma cell line.
5.5. Gliomas
Gliomas are tumors that are aggressive and there are no cures currently. Prior reports have indicated that there was a good correlation between the low expression of RKIP and a higher tumor grade [171,172]. The expression of RKIP in gliomas and its clinical significance in metastasis has been reported [150]. The findings by Martinho et al., were different from the report by Maresch et al., of the correlation between the loss of RKIP and high malignant grade [150,171]. However, the association between the loss of RKIP expression and the poor prognosis of high-grade gliomas reported by Maresch et al., was consistent with the findings of Martinho et al. [150,171]. In gliomas, RKIP expression did not affect cell proliferation, and downregulation of RKIP enhanced cell migration but did not affect tumor angiogenesis [150] in contrast to other murine cancers such as prostate [23] and breast cancers [173]. Overall, these findings suggested that the loss of RKIP expression correlates with a poor clinical outcome in glioma patients.
5.6. Renal Cell Carcinoma
Clear cell renal cell carcinoma (ccRCC) is a cancer in which there are no reliable biomarkers for either its diagnosis or its prognosis. Papale et al., reported that urine from patients with ccRCC has high levels of RKIP and phospho-RKIP that predicted cell survival and progression-free survival [174]. Down-regulation of RKIP expression has been implicated in the development and progression of renal cell carcinoma (RCC). Recently, a study of 310 RCC cases has suggested that RKIP was a significant prognostic marker because of its close correlation with progression and metastasis of RCC. Furthermore, reduced RKIP expression was related to later disease stage, larger tumor size, sarcomatoid subtype, and poor overall survival. These authors also reported that the genetic polymorphisms in RKIP might be associated with the susceptibility and progression of RCC [175].
5.7. Gastric Cancer
RKIP expression in intestinal-type gastric cancer was reported to be significantly lower and the authors proposed that RKIP is an independent prognostic factor for intestinal gastric cancer [17]. Furthermore, immunohistochemical analysis showed that the RKIP expression level was the highest in nonneoplastic gastric tissue, low in primary gastric cancer tissue, and the lowest in metastatic gastric cancer tissue, suggesting that RKIP may play a role in the tumorigenesis and metastasis of gastric cancer [176]. Additional studies revealed that RKIP protein expression was negatively correlated with the depth of invasion, TNM stage, and lymph node metastasis. Further clinical and pathological analyses revealed that RKIP protein expression was negatively correlated with the depth of invasion, TNM stage, and lymph node metastasis [132,176,177].
5.8. Lung Cancer
Lung cancer is the most common and fatal for both male and females. Lung cancer is usually divided into two main categories: small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). The latter is further divided into three major types, squamous cell carcinoma (SCC), adenocarcinoma (AC), and large cell carcinoma [178]. Lung AC is the most common type of lung cancer and has a poor survival rate. Studies on the RKIP expression and its clinical significance in lung cancer are not very conclusive. There is one study that assessed the expression levels of the inactive form of RKIP with phosphorylation at serine 153 (pRKIP) [65]. In agreement with this, studies in melanoma and breast cancer have also shown that low levels of pRKIP could predict poor survival in comparison with relatively higher expression [19,179]. Studies by our group analyzed RKIP mRNA expression across 37 different cancer types and, using data from The Cancer Genome Atlas (TCGA) platform, showed that RKIP is downregulated in lung cancer compared to normal lung tissues, with lung adenocarcinoma being among the eight tumor types with the lowest RKIP expression levels [12].
5.9. Leukemia
RKIP plays a major role in physiologic hematopoiesis and myeloid malignancies. In physiologic hematopoiesis, a decrease in RKIP expression in the HSPC pool increases the myelomonocytic lineage commitment of these cells. RKIP loss has been described in acute myeloid leukemia (AML) and a series of other myeloid neoplasia (MNs), and a functional involvement in myeloid leukemogenesis has been proven. These same authors have shown that RKIP inhibits proliferation and transformation of myeloid cells and decreases the transformation that is induced by mutant RAS. Both in vitro and in vivo experiments demonstrated that RKIP is an essential player within the development of these liquid tumors. They postulated that RKIP expression is of prognostic relevance and is a target for enhancing therapeutic strategies in AML [180].
5.10. Multiple Myeloma
Multiple myeloma (MM) is a clonal plasma-cell neoplastic disorder arising from an indolent premalignant disease known as monoclonal gammopathy of undetermined significance (MGUS). All tumors examined have exhibited low levels of RKIP; in contrast, RKIP is overexpressed primarily in its inactive phosphorylated form in MM cell lines and patient-derived tumor tissues. RKIP and the inactivated p-Ser153 form of RKIP are overexpressed in multiple myeloma cell lines and patients’ tissues compared to other tumors, healthy B cells, and healthy bone marrow. Specifically, about half of the RKIP positive cells in MM are in the phosphorylated form. The high RKIP expression in MM is positively correlated with a more aggressive diagnosis, usually resulting in a worse prognosis [90].
5.11. Other Cancers
RKIP has been identified as an important protein in various cancer types, several of which have been described above. In the majority of different types of cancers, RKIP exhibits low expression levels and RKIP is often absent in metastasis. RKIP loss has been suggested to result from the hypermethylation of its promoter. RKIP mRNA expression across 37 different cancer types was measured using data from The Cancer Genome Atlas (TCGA) platform, corroborating its downregulation in the majority of them compared to the normal tissues. This analysis showed that RKIP exhibits its highest levels in adrenocortical carcinoma (ACC), liver hepatocellular carcinoma (LIHC), and thyroid carcinoma (THCA), and its lowest expression was detected in acute myeloid leukemia (LAML), esophageal carcinoma (ESCA), and stomach and esophageal carcinomas (STES) [12].
This entry is adapted from the peer-reviewed paper 10.3390/cancers13246247
This entry is offline, you can click here to edit this entry!