Telomeres cap the ends of eukaryotic chromosomes and are indispensable chromatin structures for genome protection and replication. Telomere length maintenance has been attributed to several functional modulators, including telomerase, the shelterin complex, and the CST complex, synergizing with DNA replication, repair, and the RNA metabolism pathway components. As dysfunctional telomere maintenance and telomerase activation are associated with several human diseases, including cancer, the molecular mechanisms behind telomere length regulation and protection need particular emphasis. Cancer cells exhibit telomerase activation, enabling replicative immortality. Telomerase reverse transcriptase (TERT) activation is involved in cancer development through diverse activities other than mediating telomere elongation.
- telomerase
- telomerase reverse transcriptase
- shelterin
- CST
- promoter mutations
1. Introduction
2. Telomeres, a Genetic Time Bomb or a Biological Clock
3. The Shelterin Complex
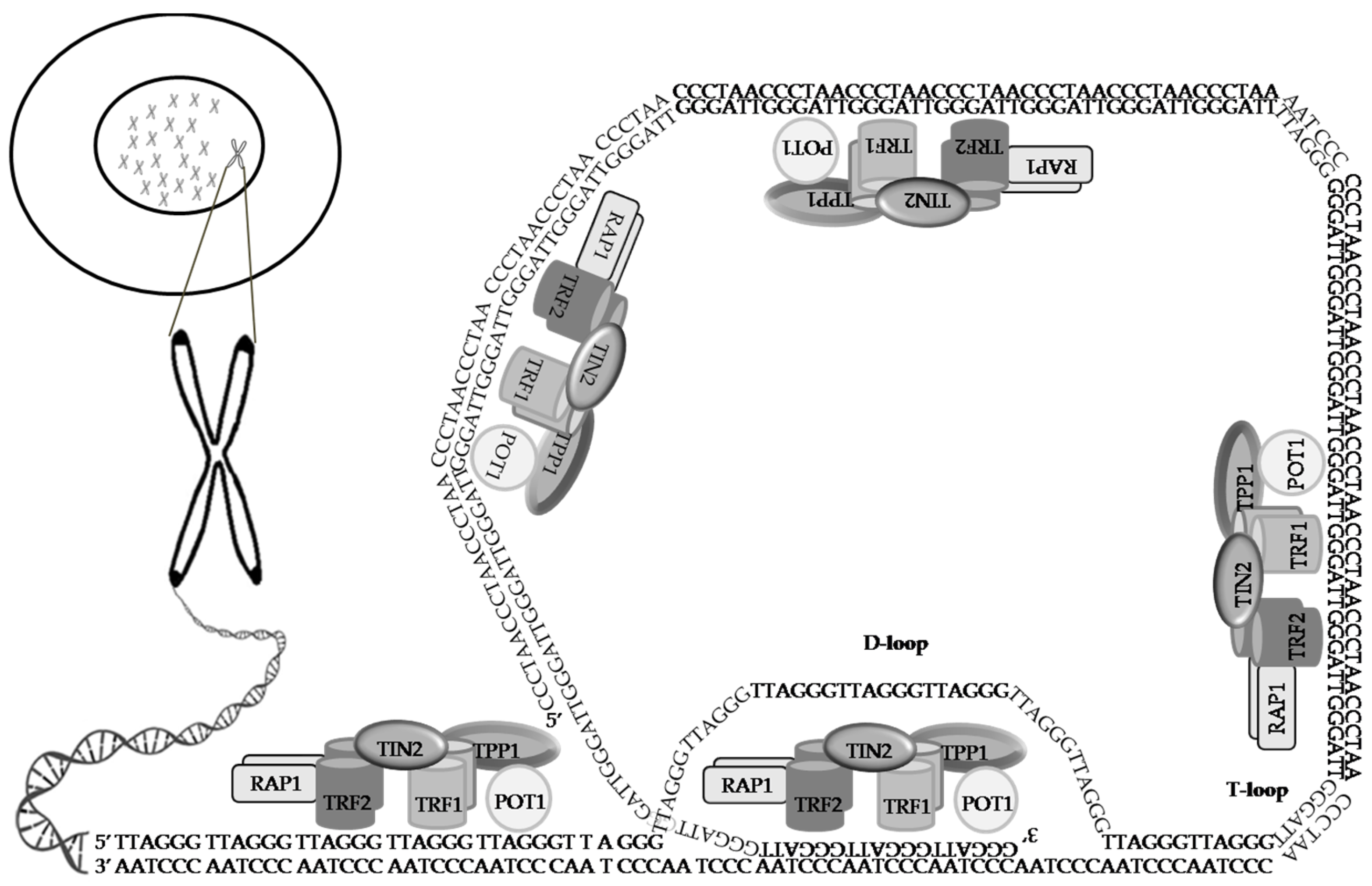
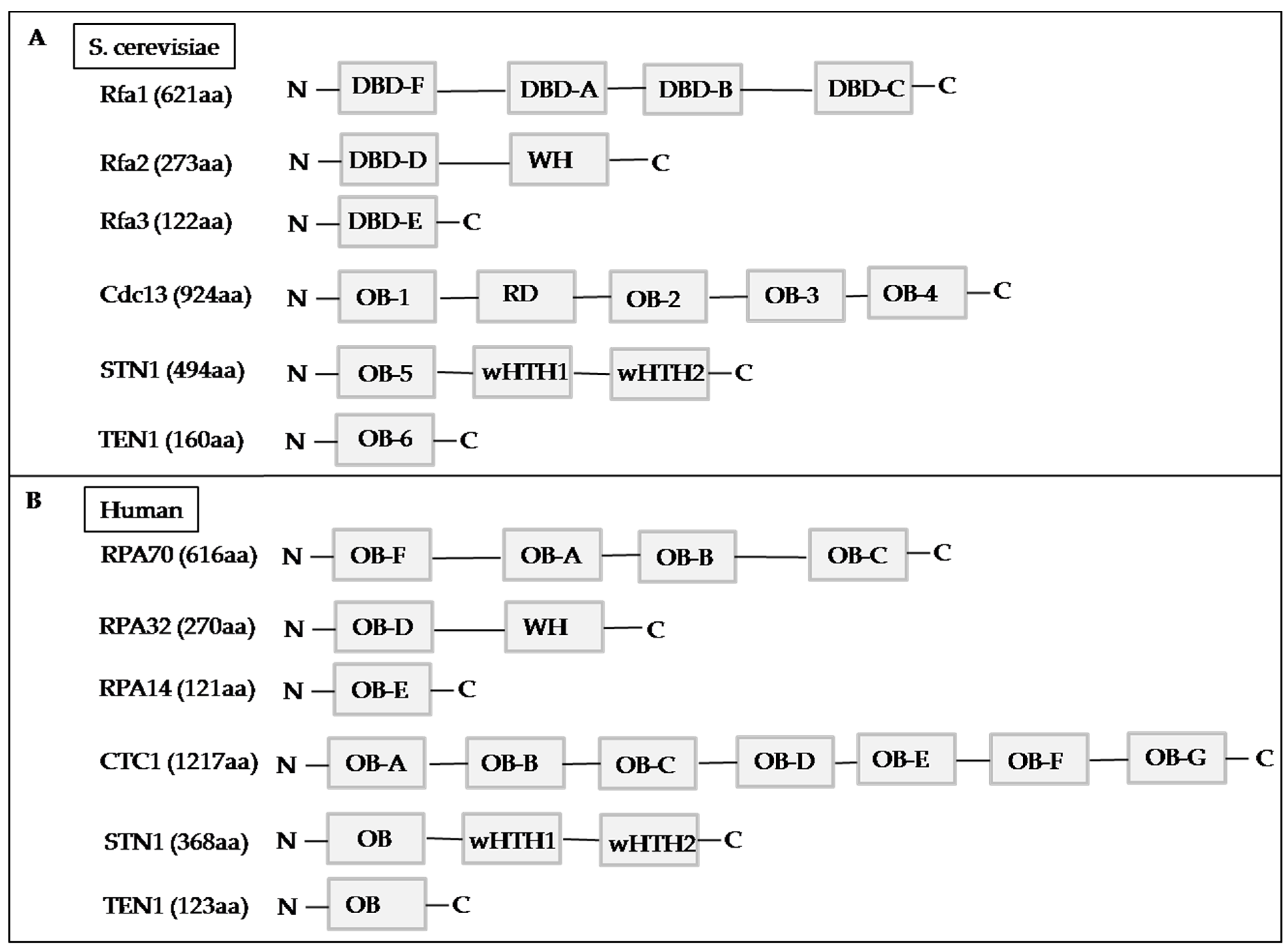
4. The CST Complex
4.1. Yeast CST Complex
4.2. Human CST Complex
|
CST. |
Component |
aa |
OB |
wH |
wHTH1 |
Functions |
References |
|---|---|---|---|---|---|---|---|
|
1. Binding to ssDNA. |
|||||||
|
CTC1 |
1217 |
7 |
0 |
0 |
|
[65] |
|
|
STN1 |
368 |
1 |
0 |
2 |
|
[66] |
|
|
TEN1 |
123 |
1 |
0 |
0 |
|
[67] |
|
|
|||||||
|
|||||||
|
[66] |
||||||
|
|||||||
|
[72] |
||||||
|
[73] |
||||||
|
|||||||
|
RPA |
|
[76] |
|||||
|
RPA70 |
616 |
4 |
0 |
0 |
|
[77] |
|
|
RPA32 |
270 |
1 |
1 |
0 |
|
[78] |
|
|
RPA14 |
121 |
1 |
0 |
0 |
|
[79] |
|
|
[80] |
||||||
|
[81] |
||||||
|
[82] |
||||||
|
[83] |
4.3. CTC1
4.4. STN1
4.5. TEN1
5. Telomerase: Breaking through the Limitation of Replication
6. Telomerase-Based Anti-Cancer Strategy
6.1. GV1001
6.2. GX301
6.3. UV1
6.4. Vx-001
7. Alternative Lengthening of Telomere (ALT)
7.1. ATRX and DAXX
7.2. Correlation between the Loss-of-Function of ATRX/DAXX and ALT in Cancer
7.3. Targeting Telomerase Activity and the ATRX/DAXX Complex
This entry is adapted from the peer-reviewed paper 10.3390/life11121405
References
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Cancer statistics for the year 2020: An overview. Int. J. Cancer 2021, 149, 778–789.
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33.
- Vogelstein, B.; Kinzler, K.W. The Path to Cancer—Three Strikes and You’re out. N. Engl. J. Med. 2015, 373, 1895–1898.
- Krupp, G.; Bonatz, G.; Parwaresch, R. Telomerase, immortality and cancer. Biotechnol. Annu. Rev. 2000, 6, 103–140.
- Cong, Y.-S.; Wright, W.E.; Shay, J.W. Human Telomerase and Its Regulation. Microbiol. Mol. Biol. Rev. 2002, 66, 407–425.
- Lim, C.J.; Cech, T.R. Shaping human telomeres: From shelterin and CST complexes to telomeric chromatin organization. Nat. Rev. Mol. Cell Biol. 2021, 22, 283–298.
- De Lange, T. How telomeres solve the end-protection problem. Science 2009, 326, 948–952.
- Meyne, J.; Ratliff, R.L.; Moyzis, R.K. Conservation of the human telomere sequence (TTAGGG)n among vertebrates. Proc. Natl. Acad. Sci. USA 1989, 86, 7049–7053.
- Lundblad, V. Telomere end processing: Unexpected complexity at the end game: Figure 1. Genes Dev. 2012, 26, 1123–1127.
- Gong, J.; Costanzo, A.; Yang, H.-Q.; Melino, G.; Kaelin, W.G., Jr.; Levrero, M.; Wang, J.Y.J. The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage. Nature 1999, 399, 806–809.
- Stiewe, T.; Putzer, B.M. P73 in apoptosis. Apoptosis 2001, 6, 447–452.
- O’Sullivan, R.J.; Karlseder, J. Telomeres: Protecting chromosomes against genome instability. Nat. Rev. Mol. Cell Biol. 2010, 11, 171–181.
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217.
- Zhao, Y.; Abreu, E.; Kim, J.; Stadler, G.; Eskiocak, U.; Terns, M.P.; Terns, R.M.; Shay, J.W.; Wright, W.E. Processive and Distributive Extension of Human Telomeres by Telomerase under Homeostatic and Nonequilibrium Conditions. Mol. Cell 2011, 42, 297–307.
- Li, B.; Reddy, S.; Comai, L. Sequence-specific processing of telomeric 3′ overhangs by the Werner syndrome protein exonuclease activity. Aging 2009, 1, 289–302.
- Muraki, K.; Nyhan, K.; Han, L.; Murnane, J.P. Mechanisms of telomere loss and their consequences for chromosome instability. Front. Oncol. 2012, 2, 135.
- Frenck, R.W., Jr.; Blackburn, E.H.; Shannon, K.M. The rate of telomere sequence loss in human leukocytes varies with age. Proc. Natl. Acad. Sci. USA 1998, 95, 5607–5610.
- Dalgård, C.; Benetos, A.; Verhulst, S.; Labat, C.; Kark, J.D.; Christensen, K.; Kimura, M.; Kyvik, K.O.; Aviv, A. Leukocyte telomere length dynamics in women and men: Menopause vs age effects. Int. J. Epidemiol. 2015, 44, 1688–1695.
- Coburn, S.B.; Graubard, B.I.; Trabert, B.; McGlynn, K.A.; Cook, M.B. Associations between circulating sex steroid hormones and leukocyte telomere length in men in the National Health and Nutrition Examination Survey. Andrology 2018, 6, 542–546.
- Arsenis, N.C.; You, T.; Ogawa, E.F.; Tinsley, G.M.; Zuo, L. Physical activity and telomere length: Impact of aging and potential mechanisms of action. Oncotarget 2017, 8, 45008–45019.
- Welendorf, C.; Nicoletti, C.F.; Pinhel, M.A.D.S.; Noronha, N.; de Paula, B.M.F.; Nonino, C.B. Obesity, weight loss, and influence on telomere length: New insights for personalized nutrition. Nutrition 2019, 66, 115–121.
- Salihu, H.M.; Pradhan, A.; King, L.; Paothong, A.; Nwoga, C.; Marty, P.J.; Whiteman, V. Impact of intrauterine tobacco exposure on fetal telomere length. Am. J. Obstet. Gynecol. 2015, 212, 205.e1–205.e8.
- Leung, C.W.; Fung, T.T.; McEvoy, C.T.; Lin, J.; Epel, E.S. Diet Quality Indices and Leukocyte Telomere Length Among Healthy US Adults: Data from the National Health and Nutrition Examination Survey, 1999–2002. Am. J. Epidemiol. 2018, 187, 2192–2201.
- Calado, R.T.; Young, N.S. Telomere diseases. N. Engl. J. Med. 2009, 361, 2353–2365.
- Jacobs, J.J.L. Loss of Telomere Protection: Consequences and Opportunities. Front. Oncol. 2013, 3, 88.
- Farzaneh-Far, R.; Cawthon, R.M.; Na, B.; Browner, W.S.; Schiller, N.B.; Whooley, M.A. Prognostic value of leukocyte telomere length in patients with stable coronary artery disease: Data from the heart and soul study. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 1379–1384.
- Yang, Z.; Huang, X.; Jiang, H.; Zhang, Y.; Liu, H.; Qin, C.; Eisner, G.M.; Jose, P.; Rudolph, L.; Ju, Z. Short Telomeres and Prognosis of Hypertension in a Chinese Population. Hypertension 2009, 53, 639–645.
- Yeh, J.-K.; Lin, M.-H.; Wang, C.-Y. Telomeres as Therapeutic Targets in Heart Disease. JACC Basic Transl. Sci. 2019, 4, 855–865.
- Sharifi-Sanjani, M.; Oyster, N.M.; Tichy, E.D.; Bedi, K.C., Jr.; Harel, O.; Margulies, K.B.; Mourkioti, F. Cardiomyocyte-Specific Telomere Shortening is a Distinct Signature of Heart Failure in Humans. J. Am. Heart Assoc. 2017, 6, e005086.
- Valdes, A.M.; Richards, J.B.; Gardner, J.P.; Swaminathan, R.; Kimura, M.; Xiaobin, L.; Aviv, A.; Spector, T.D. Telomere length in leukocytes correlates with bone mineral density and is shorter in women with osteoporosis. Osteoporos. Int. 2007, 18, 1203–1210.
- Grunnet, L.G.; Pilgaard, K.A.; Alibegovic, A.; Jensen, C.B.; Hjort, L.; Ozanne, S.; Bennett, M.; Vaag, A.A.; Brøns, C. Leukocyte telomere length is associated with elevated plasma glucose and HbA1c in young healthy men independent of birth weight. Sci. Rep. 2019, 9, 7639.
- Jafri, M.A.; Ansari, S.A.; Alqahtani, M.H.; Shay, J.W. Roles of telomeres and telomerase in cancer, and advances in telomerase-targeted therapies. Genome Med. 2016, 8, 69.
- De Lange, T. Shelterin: The protein complex that shapes and safeguards human telomeres. Genes Dev. 2005, 19, 2100–2110.
- Diotti, R.; Loayza, D. Shelterin complex and associated factors at human telomeres. Nucleus 2011, 2, 119–135.
- Chen, L.-Y.; Liu, D.; Songyang, Z. Telomere Maintenance through Spatial Control of Telomeric Proteins. Mol. Cell. Biol. 2007, 27, 5898–5909.
- Liu, D.; O’Connor, M.S.; Qin, J.; Songyang, Z. Telosome, a Mammalian Telomere-associated Complex Formed by Multiple Telomeric Proteins. J. Biol. Chem. 2004, 279, 51338–51342.
- O’Connor, M.S.; Safari, A.; Xin, H.; Liu, D.; Songyang, Z. A critical role for TPP1 and TIN2 interaction in high-order telomeric complex assembly. Proc. Natl. Acad. Sci. USA 2006, 103, 11874–11879.
- Ye, J.Z.; de Lange, T. Tin2 is a tankyrase 1 parp modulator in the trf1 telomere length control complex. Nat. Genet. 2004, 36, 618–623.
- Li, J.S.Z.; Fusté, J.M.; Simavorian, T.; Bartocci, C.; Tsai, J.; Karlseder, J.; Denchi, E.L. TZAP: A telomere-associated protein involved in telomere length control. Science 2017, 355, 638–641.
- Marcand, S.; Gilson, E.; Shore, D. A Protein-Counting Mechanism for Telomere Length Regulation in Yeast. Science 1997, 275, 986–990.
- Patel, T.N.; Vasan, R.; Gupta, D.; Patel, J.; Trivedi, M. Shelterin Proteins and Cancer. Asian Pac. J. Cancer Prev. 2015, 16, 3085–3090.
- Palm, W.; de Lange, T. How shelterin protects mammalian telomeres. Annu. Rev. Genet. 2008, 42, 301–334.
- Xin, H.; Liu, D.; Songyang, Z. The telosome/shelterin complex and its functions. Genome Biol. 2008, 9, 232.
- Tejera, A.M.; Stagno d’Alcontres, M.; Thanasoula, M.; Marion, R.M.; Martinez, P.; Liao, C.; Flores, J.M.; Tarsounas, M.; Blasco, M.A. Tpp1 is required for tert recruitment, telomere elongation during nuclear reprogramming, and normal skin development in mice. Dev. Cell 2010, 18, 775–789.
- Gao, H.; Cervantes, R.B.; Mandell, E.K.; Otero, J.H.; Lundblad, V. RPA-like proteins mediate yeast telomere function. Nat. Struct. Mol. Biol. 2007, 14, 208–214.
- Wellinger, R.J. The CST Complex and Telomere Maintenance: The Exception Becomes the Rule. Mol. Cell 2009, 36, 168–169.
- Chen, L.-Y.; Lingner, J. CST for the grand finale of telomere replication. Nucleus 2013, 4, 277–282.
- Martín, V.; Du, L.-L.; Rozenzhak, S.; Russell, P. Protection of telomeres by a conserved Stn1 Ten1 complex. Proc. Natl. Acad. Sci. USA 2007, 104, 14038–14043.
- Chen, H.-W.; Xue, J.; Churikov, D.; Hass, E.P.; Shi, S.; Lemon, L.D.; Luciano, P.; Bertuch, A.A.; Zappulla, D.C.; Géli, V.; et al. Structural Insights into Yeast Telomerase Recruitment to Telomeres. Cell 2018, 172, 331–343.e13.
- Wold, M.S. Replication Protein A: A Heterotrimeric, Single-Stranded DNA-Binding Protein Required for Eukaryotic DNA Metabolism. Annu. Rev. Biochem. 1997, 66, 61–92.
- Lim, C.J.; Barbour, A.T.; Zaug, A.J.; Goodrich, K.J.; McKay, A.E.; Wuttke, D.S.; Cech, T.R. The structure of human CST reveals a decameric assembly bound to telomeric DNA. Science 2020, 368, 1081–1085.
- Miyake, Y.; Nakamura, M.; Nabetani, A.; Shimamura, S.; Tamura, M.; Yonehara, S.; Saito, M.; Ishikawa, F. RPA-like Mammalian Ctc1-Stn1-Ten1 Complex Binds to Single-Stranded DNA and Protects Telomeres Independently of the Pot1 Pathway. Mol. Cell 2009, 36, 193–206.
- Rice, C.; Skordalakes, E. Structure and function of the telomeric CST complex. Comput. Struct. Biotechnol. J. 2016, 14, 161–167.
- Chastain, M.; Zhou, Q.; Shiva, O.; Fadri-Moskwik, M.; Whitmore, L.; Jia, P.; Dai, X.; Huang, C.; Ye, P.; Chai, W. Human CST Facilitates Genome-wide RAD51 Recruitment to GC-Rich Repetitive Sequences in Response to Replication Stress. Cell Rep. 2016, 16, 1300–1314.
- Price, C.; Boltz, K.A.; Chaiken, M.F.; Stewart, J.A.; Beilstein, M.A.; Shippen, D.E. Evolution of CST function in telomere maintenance. Cell Cycle 2010, 9, 3177–3185.
- Stewart, J.A.; Wang, Y.; Ackerson, S.M.; Schuck, P.L. Emerging roles of cst in maintaining genome stability and human disease. Front. Biosci. 2018, 23, 1564–1586.
- Maréchal, A.; Zou, L. RPA-coated single-stranded DNA as a platform for post-translational modifications in the DNA damage response. Cell Res. 2015, 25, 9–23.
- Stewart, J.A.; Wang, F.; Chaiken, M.F.; Kasbek, C.; Chastain, P.D., 2nd; Wright, W.E.; Price, C.M. Human CST promotes telomere duplex replication and general replication restart after fork stalling. EMBO J. 2012, 31, 3537–3549.
- Fanning, E.; Klimovich, V.; Nager, A.R. A dynamic model for replication protein A (RPA) function in DNA processing pathways. Nucleic Acids Res. 2006, 34, 4126–4137.
- Gu, P.; Jia, S.; Takasugi, T.; Smith, E.; Nandakumar, J.; Hendrickson, E.; Chang, S. CTC1-STN1 coordinates G- and C-strand synthesis to regulate telomere length. Aging Cell 2018, 17, e12783.
- Gu, P.; Min, J.-N.; Wang, Y.; Huang, C.; Peng, T.; Chai, W.; Chang, S. CTC1 deletion results in defective telomere replication, leading to catastrophic telomere loss and stem cell exhaustion. EMBO J. 2012, 31, 2309–2321.
- Hom, R.A.; Wuttke, D.S. Human CST Prefers G-Rich but Not Necessarily Telomeric Sequences. Biochemistry 2017, 56, 4210–4218.
- Huang, C.; Dai, X.; Chai, W. Human Stn1 protects telomere integrity by promoting efficient lagging-strand synthesis at telomeres and mediating C-strand fill-in. Cell Res. 2012, 22, 1681–1695.
- Huang, C.; Jia, P.; Chastain, M.; Shiva, O.; Chai, W. The human ctc1/stn1/ten1 complex regulates telomere maintenance in alt cancer cells. Exp. Cell Res. 2017, 355, 95–104.
- Feng, X.; Hsu, S.-J.; Bhattacharjee, A.; Wang, Y.; Diao, J.; Price, C.M. CTC1-STN1 terminates telomerase while STN1-TEN1 enables C-strand synthesis during telomere replication in colon cancer cells. Nat. Commun. 2018, 9, 2827.
- Bhattacharjee, A.; Wang, Y.; Diao, J.; Price, C.M. Dynamic DNA binding, junction recognition and G4 melting activity underlie the telomeric and genome-wide roles of human CST. Nucleic Acids Res. 2017, 45, 12311–12324.
- Boltz, K.A.; Leehy, K.; Song, X.; Nelson, A.; Shippen, D.E. ATR cooperates with CTC1 and STN1 to maintain telomeres and genome integrity in Arabidopsis. Mol. Biol. Cell 2012, 23, 1558–1568.
- Casteel, D.E.; Zhuang, S.; Zeng, Y.; Perrino, F.W.; Boss, G.R.; Goulian, M.; Pilz, R.B. A DNA polymerase-α·primase cofactor with homology to replication protein a-32 regulates DNA replication in mammalian cells. J. Biol. Chem. 2009, 284, 5807–5818.
- Ganduri, S.; Lue, N.F. STN1–POLA2 interaction provides a basis for primase-pol α stimulation by human STN1. Nucleic Acids Res. 2017, 45, 9455–9466.
- Wang, F.; Stewart, J.A.; Kasbek, C.; Zhao, Y.; Wright, W.E.; Price, C.M. Human CST Has Independent Functions during Telomere Duplex Replication and C-Strand Fill-In. Cell Rep. 2012, 2, 1096–1103.
- Chen, L.-Y.; Redon, S.; Lingner, J. The human CST complex is a terminator of telomerase activity. Nature 2012, 488, 540–544.
- Wang, Y.; Brady, K.S.; Caiello, B.P.; Ackerson, S.M.; Stewart, J.A. Human cst suppresses origin licensing and promotes and-1/ctf4 chromatin association. Life Sci. Alliance 2019, 2.
- Li, Y.; Xiao, H.; de Renty, C.; Jaramillo-Lambert, A.; Han, Z.; DePamphilis, M.L.; Brown, K.; Zhu, W. The Involvement of Acidic Nucleoplasmic DNA-binding Protein (And-1) in the Regulation of Prereplicative Complex (pre-RC) Assembly in Human Cells. J. Biol. Chem. 2012, 287, 42469–42479.
- Feng, X.; Hsu, S.-J.; Kasbek, C.; Chaiken, M.; Price, C.M. CTC1-mediated C-strand fill-in is an essential step in telomere length maintenance. Nucleic Acids Res. 2017, 45, 4281–4293.
- Wu, P.; Takai, H.; de Lange, T. Telomeric 3′ Overhangs Derive from Resection by Exo1 and Apollo and Fill-In by POT1b-Associated CST. Cell 2012, 150, 39–52.
- Feng, S.; Zhao, Y.; Xu, Y.; Ning, S.; Huo, W.; Hou, M.; Gao, G.; Ji, J.; Guo, R.; Xu, D. Ewing Tumor-associated Antigen 1 Interacts with Replication Protein A to Promote Restart of Stalled Replication Forks. J. Biol. Chem. 2016, 291, 21956–21962.
- Zeman, M.K.; Cimprich, K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014, 16, 2–9.
- Cimprich, K.A.; Cortez, D. ATR: An essential regulator of genome integrity. Nat. Rev. Mol. Cell Biol. 2008, 9, 616–627.
- Patil, M.; Pabla, N.; Dong, Z. Checkpoint kinase 1 in DNA damage response and cell cycle regulation. Cell. Mol. Life Sci. 2013, 70, 4009–4021.
- Cunniff, C.; Bassetti, J.A.; Ellis, N.A. Bloom’s syndrome: Clinical spectrum, molecular pathogenesis, and cancer predisposition. Mol. Syndromol. 2017, 8, 4–23.
- Guler, G.D.; Liu, H.; Vaithiyalingam, S.; Arnett, D.R.; Kremmer, E.; Chazin, W.J.; Fanning, E. Human DNA Helicase B (HDHB) Binds to Replication Protein A and Facilitates Cellular Recovery from Replication Stress. J. Biol. Chem. 2012, 287, 6469–6481.
- Lee, M.; Shin, S.; Uhm, H.; Hong, H.; Kirk, J.; Hyun, K.; Kulikowicz, T.; Kim, J.; Ahn, B.; Bohr, V.A.; et al. Multiple RPAs make WRN syndrome protein a superhelicase. Nucleic Acids Res. 2018, 46, 4689–4698.
- Qin, Z.; Bi, L.; Hou, X.-M.; Zhang, S.; Zhang, X.; Lu, Y.; Li, M.; Modesti, M.; Xi, X.-G.; Sun, B. Human RPA activates BLM’s bidirectional DNA unwinding from a nick. eLife 2020, 9, 9.
- Bhat, K.; Bétous, R.; Cortez, D. High-affinity DNA-binding Domains of Replication Protein A (RPA) Direct SMARCAL1-dependent Replication Fork Remodeling. J. Biol. Chem. 2015, 290, 4110–4117.
- Martínez-Jiménez, M.I.; Lahera, A.; Blanco, L. Human PrimPol activity is enhanced by RPA. Sci. Rep. 2017, 7, 783.
- Garvik, B.; Carson, M.; Hartwell, L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol. Cell. Biol. 1995, 15, 6128–6138.
- Bryan, C.; Rice, C.; Harkisheimer, M.; Schultz, D.C.; Skordalakes, E. Structure of the Human Telomeric Stn1-Ten1 Capping Complex. PLoS ONE 2013, 8, e66756.
- Hughes, T.R.; Weilbaecher, R.G.; Walterscheid, M.; Lundblad, V. Identification of the single-strand telomeric DNA binding domain of the Saccharomyces cerevisiae Cdc13 protein. Proc. Natl. Acad. Sci. USA 2000, 97, 6457–6462.
- Lewis, K.A.; Pfaff, D.A.; Earley, J.N.; Altschuler, S.E.; Wuttke, D.S. The tenacious recognition of yeast telomere sequence by Cdc13 is fully exerted by a single OB-fold domain. Nucleic Acids Res. 2014, 42, 475–484.
- Sun, J.; Yang, Y.; Wan, K.; Mao, N.; Yu, T.-Y.; Lin, Y.-C.; DeZwaan, D.C.; Freeman, B.C.; Lin, J.-J.; Lue, N.F.; et al. Structural bases of dimerization of yeast telomere protein Cdc13 and its interaction with the catalytic subunit of DNA polymerase α. Cell Res. 2010, 21, 258–274.
- Ge, Y.; Wu, Z.; Chen, H.; Zhong, Q.; Shi, S.; Li, G.; Wu, J.; Lei, M. Structural insights into telomere protection and homeostasis regulation by yeast CST complex. Nat. Struct. Mol. Biol. 2020, 27, 752–762.
- Bochkareva, E.; Korolev, S.; Lees-Miller, S.P.; Bochkarev, A. Structure of the RPA trimerization core and its role in the multistep DNA-binding mechanism of RPA. EMBO J. 2002, 21, 1855–1863.
- Li, S.; Makovets, S.; Matsuguchi, T.; Blethrow, J.D.; Shokat, K.M.; Blackburn, E.H. Cdk1-Dependent Phosphorylation of Cdc13 Coordinates Telomere Elongation during Cell-Cycle Progression. Cell 2009, 136, 50–61.
- Liu, C.-C.; Gopalakrishnan, V.; Poon, L.-F.; Yan, T.; Li, S. Cdk1 Regulates the Temporal Recruitment of Telomerase and Cdc13-Stn1-Ten1 Complex for Telomere Replication. Mol. Cell. Biol. 2014, 34, 57–70.
- Tseng, S.-F.; Shen, Z.-J.; Tsai, H.-J.; Lin, Y.-H.; Teng, S.-C. Rapid Cdc13 turnover and telomere length homeostasis are controlled by Cdk1-mediated phosphorylation of Cdc13. Nucleic Acids Res. 2009, 37, 3602–3611.
- Wu, Y.; DiMaggio, P.A., Jr.; Perlman, D.H.; Zakian, V.A.; Garcia, B.A. Novel Phosphorylation Sites in the S. cerevisiae Cdc13 Protein Reveal New Targets for Telomere Length Regulation. J. Proteome Res. 2013, 12, 316–327.
- Zhang, W.; Durocher, D. De novo telomere formation is suppressed by the Mec1-dependent inhibition of Cdc13 accumulation at DNA breaks. Genes Dev. 2010, 24, 502–515.
- Poncet, D.; Belleville, A.; de Roodenbeke, C.T.; de Climens, A.R.; Ben Simon, E.; Merle-Beral, H.; Callet-Bauchu, E.; Salles, G.; Sabatier, L.; Delic, J.; et al. Changes in the expression of telomere maintenance genes suggest global telomere dysfunction in B-chronic lymphocytic leukemia. Blood 2008, 111, 2388–2391.
- Chandra, A.; Hughes, T.R.; Nugent, C.I.; Lundblad, V. Cdc13 both positively and negatively regulates telomere replication. Genes Dev. 2001, 15, 404–414.
- Evans, S.K.; Lundblad, V. Positive and negative regulation of telomerase access to the telomere. J. Cell Sci. 2000, 113, 3357–3364.
- Zhong, F.; Batista, L.; Freund, A.; Pech, M.F.; Venteicher, A.; Artandi, S.E. TPP1 OB-Fold Domain Controls Telomere Maintenance by Recruiting Telomerase to Chromosome Ends. Cell 2012, 150, 481–494.
- Giraud-Panis, M.-J.; Teixeira, M.T.; Géli, V.; Gilson, E. CST Meets Shelterin to Keep Telomeres in Check. Mol. Cell 2010, 39, 665–676.
- Langston, R.E.; Palazzola, D.; Bonnell, E.; Wellinger, R.J.; Weinert, T. Loss of Cdc13 causes genome instability by a deficiency in replication-dependent telomere capping. PLoS Genet. 2020, 16, e1008733.
- Han, E.; Patel, N.A.; Yannuzzi, N.A.; Laura, D.M.; Fan, K.C.; Negron, C.I.; Prakhunhungsit, S.; Thorson, W.L.; Berrocal, A.M. A unique case of coats plus syndrome and dyskeratosis congenita in a patient with CTC1 mutations. Ophthalmic Genet. 2020, 41, 363–367.
- Bs, R.B.K.; Bs, K.E.G.; Usmani, G.N.; Asdourian, G.K.; Williams, D.A.; Hofmann, I.; Agarwal, S. CTC1 Mutations in a patient with dyskeratosis congenita. Pediatr. Blood Cancer 2012, 59, 311–314.
- Dai, X.; Huang, C.; Bhusari, A.; Sampathi, S.; Schubert, K.; Chai, W. Molecular steps of G-overhang generation at human telomeres and its function in chromosome end protection. EMBO J. 2010, 29, 2788–2801.
- Wang, Y.; Chai, W. Pathogenic CTC1 mutations cause global genome instabilities under replication stress. Nucleic Acids Res. 2018, 46, 3981–3992.
- Goulian, M.; Heard, C.J. The mechanism of action of an accessory protein for DNA polymerase alpha/primase. J. Biol. Chem. 1990, 265, 13231–13239.
- Grossi, S.; Puglisi, A.; Dmitriev, P.V.; Lopes, M.; Shore, D. Pol12, the B subunit of DNA polymerase α, functions in both telomere capping and length regulation. Genes Dev. 2004, 18, 992–1006.
- Petreaca, R.C.; Chiu, H.-C.; Eckelhoefer, H.A.; Chuang, C.; Xu, L.; Nugent, C.I. Chromosome end protection plasticity revealed by Stn1p and Ten1p bypass of Cdc13p. Nat. Cell Biol. 2006, 8, 748–755.
- Pennock, E.; Buckley, K.; Lundblad, V. Cdc13 Delivers Separate Complexes to the Telomere for End Protection and Replication. Cell 2001, 104, 387–396.
- Qian, W.; Wang, J.; Jin, N.-N.; Fu, X.-H.; Lin, Y.-C.; Lin, J.-J.; Zhou, J.-Q. Ten1p promotes the telomeric DNA-binding activity of Cdc13p: Implication for its function in telomere length regulation. Cell Res. 2009, 19, 849–863.
- Wang, F.; Stewart, J.; Price, C.M. Human cst abundance determines recovery from diverse forms of DNA damage and replication stress. Cell Cycle 2014, 13, 3488–3498.
- Bagcchi, S. POT1: A genetic link for familial glioma. Lancet Oncol. 2015, 16, e12.
- Robles-Espinoza, C.D.; Harland, M.; Ramsay, A.J.; Aoude, L.G.; Quesada, V.; Ding, Z.; Pooley, K.A.; Pritchard, A.L.; Tiffen, J.C.; Petljak, M.; et al. POT1 loss-of-function variants predispose to familial melanoma. Nat. Genet. 2014, 46, 478–481.
- Ramsay, A.J.; Quesada, V.; Foronda, M.; Conde, L.; Martínez-Trillos, A.; Villamor, N.; Rodríguez, D.; Kwarciak, A.; Garabaya, C.; Gallardo, M.; et al. POT1 mutations cause telomere dysfunction in chronic lymphocytic leukemia. Nat. Genet. 2013, 45, 526–530.
- Calvete, O.; Martinez, P.; Garcia-Pavia, P.; Benitez-Buelga, C.; Paumard-Hernandez, B.; Fernandez, V.; Dominguez, F.; Salas, C.; Romero-Laorden, N.; Garcia-Donas, J.; et al. A mutation in the POT1 gene is responsible for cardiac angiosarcoma in TP53-negative Li–Fraumeni-like families. Nat. Commun. 2015, 6, 8383.
- Shen, J.; Gammon, M.D.; Wu, H.-C.; Terry, M.B.; Wang, Q.; Bradshaw, P.T.; Teitelbaum, S.L.; Neugut, A.I.; Santella, R.M. Multiple Genetic Variants in Telomere Pathway Genes and Breast Cancer Risk. Cancer Epidemiol. Biomark. Prev. 2010, 19, 219–228.
- Richard, M.A.; Lupo, P.J.; Morton, L.M.; Yasui, Y.A.; Sapkota, Y.A.; Arnold, M.A.; Aubert, G.; Neglia, J.P.; Turcotte, L.M.; Leisenring, W.M.; et al. Genetic variation in POT1 and risk of thyroid subsequent malignant neoplasm: A report from the Childhood Cancer Survivor Study. PLoS ONE 2020, 15, e0228887.
- Hiyama, E.; Hiyama, K. Telomere and telomerase in stem cells. Br. J. Cancer 2007, 96, 1020–1024.
- Lucibello, F.; Menegatti, S.; Menger, L. Methods to edit T cells for cancer immunotherapy. Methods Enzymol. 2020, 631, 107–135.
- Borah, S.; Xi, L.; Zaug, A.J.; Powell, N.M.; Dancik, G.M.; Cohen, S.B.; Costello, J.C.; Theodorescu, D.; Cech, T.R. TERTpromoter mutations and telomerase reactivation in urothelial cancer. Science 2015, 347, 1006–1010.
- Inderberg-Suso, E.-M.; Trachsel, S.; Lislerud, K.; Rasmussen, A.-M.; Gaudernack, G. Widespread CD4+ T-cell reactivity to novel hTERT epitopes following vaccination of cancer patients with a single hTERT peptide GV1001. OncoImmunology 2012, 1, 670–686.
- Brunsvig, P.F.; Aamdal, S.; Gjertsen, M.K.; Kvalheim, G.; Markowski-Grimsrud, C.J.; Sve, I.; Dyrhaug, M.; Trachsel, S.; Møller, M.; Eriksen, J.A.; et al. Telomerase peptide vaccination: A phase I/II study in patients with non-small cell lung cancer. Cancer Immunol. Immunother. 2006, 55, 1553–1564.
- Greten, T.F.; Forner, A.; Korangy, F.; N’Kontchou, G.; Barget, N.; Ayuso, C.; Ormandy, L.A.; Manns, M.P.; Beaugrand, M.; Bruix, J. A phase II open label trial evaluating safety and efficacy of a telomerase peptide vaccination in patients with advanced hepatocellular carcinoma. BMC Cancer 2010, 10, 209.
- Hunger, R.E.; Lang, K.K.; Markowski, C.J.; Trachsel, S.; Møller, M.; Eriksen, J.A.; Rasmussen, A.-M.; Braathen, L.R.; Gaudernack, G. Vaccination of patients with cutaneous melanoma with telomerase-specific peptides. Cancer Immunol. Immunother. 2011, 60, 1553–1564.
- Kyte, J.A.; Gaudernack, G.; Dueland, S.; Trachsel, S.; Julsrud, L.; Aamdal, S. Telomerase Peptide Vaccination Combined with Temozolomide: A Clinical Trial in Stage IV Melanoma Patients. Clin. Cancer Res. 2011, 17, 4568–4580.
- Middleton, G.; Silcocks, P.; Cox, T.; Valle, J.; Wadsley, J.; Propper, D.; Coxon, F.; Ross, P.; Madhusudan, S.; Roques, T.; et al. Gemcitabine and capecitabine with or without telomerase peptide vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer (TeloVac): An open-label, randomised, phase 3 trial. Lancet Oncol. 2014, 15, 829–840.
- Mizukoshi, E.; Kaneko, S. Telomerase-Targeted Cancer Immunotherapy. Int. J. Mol. Sci. 2019, 20, 1823.
- Staff, C.; Mozaffari, F.; Frodin, J.-E.; Mellstedt, H.; Liljefors, M.G. Telomerase (GV1001) vaccination together with gemcitabine in advanced pancreatic cancer patients. Int. J. Oncol. 2014, 45, 1293–1303.
- Khong, H.; Overwijk, W.W. Adjuvants for peptide-based cancer vaccines. J. Immunother. Cancer 2016, 4, 56.
- Kim, B.-K.; Kim, B.-R.; Lee, H.-J.; Lee, S.-A.; Kim, B.-J.; Kim, H.; Won, Y.-S.; Shon, W.-J.; Lee, N.-R.; Inn, K.-S.; et al. Tumor-suppressive effect of a telomerase-derived peptide by inhibiting hypoxia-induced HIF-1α-VEGF signaling axis. Biomaterials 2014, 35, 2924–2933.
- Kim, C.; Lee, S.G.; Yang, W.M.; Arfuso, F.; Um, J.Y.; Kumar, A.P.; Bian, J.; Sethi, G.; Ahn, K.S. Formononetin-induced oxidative stress abrogates the activation of stat3/5 signaling axis and suppresses the tumor growth in multiple myeloma preclinical model. Cancer Lett. 2018, 431, 123–141.
- Kim, G.E.; Jung, A.R.; Kim, M.Y.; Lee, J.B.; Im, J.H.; Lee, K.W.; Park, Y.H.; Lee, J.Y. GV1001 Induces Apoptosis by Reducing Angiogenesis in Renal Cell Carcinoma Cells Both In Vitro and In Vivo. Urology 2018, 113, 129–137.
- Park, Y.H.; Jung, A.R.; Kim, G.E.; Kim, M.Y.; Sung, J.W.; Shin, D.; Cho, H.J.; Ha, U.S.; Hong, S.H.; Kim, S.W.; et al. Gv1001 inhibits cell viability and induces apoptosis in castration-resistant prostate cancer cells through the akt/nf-kappab/vegf pathway. J. Cancer 2019, 10, 6269–6277.
- Kim, H.; Seo, E.-H.; Lee, S.-H.; Kim, B.-J. The Telomerase-Derived Anticancer Peptide Vaccine GV1001 as an Extracellular Heat Shock Protein-Mediated Cell-Penetrating Peptide. Int. J. Mol. Sci. 2016, 17, 2054.
- Schlapbach, C.; Yerly, D.; Daubner, B.; Yawalkar, N.; Hunger, R.E. Telomerase-specific GV1001 peptide vaccination fails to induce objective tumor response in patients with cutaneous T cell lymphoma. J. Dermatol. Sci. 2011, 62, 75–83.
- Fenoglio, D.; Traverso, P.; Parodi, A.; Tomasello, L.; Negrini, S.; Kalli, F.; Battaglia, F.; Ferrera, F.; Sciallero, M.S.; Murdaca, G.; et al. A multi-peptide, dual-adjuvant telomerase vaccine (GX301) is highly immunogenic in patients with prostate and renal cancer. Cancer Immunol. Immunother. 2013, 62, 1041–1052.
- Aucouturier, J.; Dupuis, L.; Deville, S.; Ascarateil, S.; Ganne, V. Montanide ISA 720 and 51: A new generation of water in oil emulsions as adjuvants for human vaccines. Expert Rev. Vaccines 2002, 1, 111–118.
- Johnston, D.; Bystryn, J.-C. Topical imiquimod is a potent adjuvant to a weakly-immunogenic protein prototype vaccine. Vaccine 2006, 24, 1958–1965.
- Fenoglio, D.; Parodi, A.; Lavieri, R.; Kalli, F.; Ferrera, F.; Tagliamacco, A.; Guastalla, A.; Lamperti, M.G.; Giacomini, M.; Filaci, G. Immunogenicity of GX301 cancer vaccine: Four (telomerase peptides) are better than one. Hum. Vaccines Immunother. 2015, 11, 838–850.
- Filaci, G.; Fenoglio, D.; Nolè, F.; Zanardi, E.; Tomasello, L.; Aglietta, M.; Del Conte, G.; Carles, J.; Morales-Barrera, R.; Guglielmini, P.; et al. Telomerase-based GX301 cancer vaccine in patients with metastatic castration-resistant prostate cancer: A randomized phase II trial. Cancer Immunol. Immunother. 2021, 70, 1–14.
- Van der Burg, S.H. Correlates of immune and clinical activity of novel cancer vaccines. Semin. Immunol. 2018, 39, 119–136.
- Ellingsen, E.B.; Aamdal, E.; Inderberg, E.M.; Rasch, W.; Brunsvig, P.; Aamdal, S.; Hovig, E.; Nyakas, M.; Guren, T.K.; Gaudernack, G. A phase I/IIa clinical trial investigating the therapeutic cancer vaccine UV1 in combination with ipilimumab in patients with malignant melanoma: Four-year survival update. J. Clin. Oncol. 2020, 38, 62.
- Haakensen, V.D.; Nowak, A.K.; Ellingsen, E.B.; Farooqi, S.J.; Bjaanæs, M.M.; Horndalsveen, H.; Mcculloch, T.; Grundberg, O.; Cedres, S.M.; Helland, Å. NIPU: A randomised, open-label, phase II study evaluating nivolumab and ipilimumab combined with UV1 vaccination as second line treatment in patients with malignant mesothelioma. J. Transl. Med. 2021, 19, 1–9.
- Zakharia, Y.; O’Day, S.; Rasch, W.; Milhem, M.M. A phase I clinical trial investigating the telomerase vaccine UV1 in combination with pembrolizumab in patients with advanced melanoma. J. Clin. Oncol. 2021, 39, 2620.
- Vetsika, E.-K.; Papadimitraki, E.; Aggouraki, D.; Konsolakis, G.; Mela, M.-E.; Kotsakis, A.; Christou, S.; Patramani, S.; Alefantinou, M.; Kaskara, A.; et al. Sequential Administration of the Native TERT572 Cryptic Peptide Enhances the Immune Response Initiated by its Optimized Variant TERT572Y in Cancer Patients. J. Immunother. 2011, 34, 641–650.
- Bolonaki, I.; Kotsakis, A.; Papadimitraki, E.; Aggouraki, D.; Konsolakis, G.; Vagia, A.; Christophylakis, C.; Nikoloudi, I.; Magganas, E.; Galanis, A.; et al. Vaccination of Patients with Advanced Non–Small-Cell Lung Cancer With an Optimized Cryptic Human Telomerase Reverse Transcriptase Peptide. J. Clin. Oncol. 2007, 25, 2727–2734.
- Kotsakis, A.; Papadimitraki, E.; Vetsika, E.K.; Aggouraki, D.; Dermitzaki, E.K.; Hatzidaki, D.; Kentepozidis, N.; Mavroudis, D.; Georgoulias, V. A phase II trial evaluating the clinical and immunologic response of HLA-A2+ non-small cell lung cancer patients vaccinated with an hTERT cryptic peptide. Lung Cancer 2014, 86, 59–66.
- Kotsakis, A.; Vetsika, E.-K.; Christou, S.; Hatzidaki, D.; Vardakis, N.; Aggouraki, D.; Konsolakis, G.; Georgoulias, V.; Christophyllakis, C.; Cordopatis, P.; et al. Clinical outcome of patients with various advanced cancer types vaccinated with an optimized cryptic human telomerase reverse transcriptase (TERT) peptide: Results of an expanded phase II study. Ann. Oncol. 2012, 23, 442–449.
- Vetsika, E.-K.; Konsolakis, G.; Aggouraki, D.; Kotsakis, A.; Papadimitraki, E.; Christou, S.; Menez-Jamet, J.; Kosmatopoulos, K.; Georgoulias, V.; Mavroudis, D. Immunological responses in cancer patients after vaccination with the therapeutic telomerase-specific vaccine Vx-001. Cancer Immunol. Immunother. 2012, 61, 157–168.
- Brower, V. Telomerase-Based Therapies Emerging Slowly. J. Natl. Cancer Inst. 2010, 102, 520–521.
- Ruden, M.; Puri, N. Novel anticancer therapeutics targeting telomerase. Cancer Treat. Rev. 2013, 39, 444–456.
- Salazar-Onfray, F.; Pereda, C.; Reyes, D.; López, M.N. TAPCells, the Chilean dendritic cell vaccine against melanoma and prostate cancer. Biol. Res. 2013, 46, 431–440.
- Yan, J.; Pankhong, P.; Shin, T.H.; Obeng-Adjei, N.; Morrow, M.P.; Walters, J.N.; Khan, A.S.; Sardesai, N.; Weiner, D.B. Highly Optimized DNA Vaccine Targeting Human Telomerase Reverse Transcriptase Stimulates Potent Antitumor Immunity. Cancer Immunol. Res. 2013, 1, 179–189.
- Thalmensi, J.; Pliquet, E.; Liard, C.; Escande, M.; Bestetti, T.; Julithe, M.; Kostrzak, A.; Pailhes-Jimenez, A.-S.; Bourges, E.; Loustau, M.; et al. Anticancer DNA vaccine based on human telomerase reverse transcriptase generates a strong and specific T cell immune response. OncoImmunology 2016, 5, e1083670.
- Aurisicchio, L.; Fridman, A.; Mauro, D.; Sheloditna, R.; Chiappori, A.; Bagchi, A.; Ciliberto, G. Safety, tolerability and immunogenicity of v934/v935 htert vaccination in cancer patients with selected solid tumors: A phase I study. J. Transl. Med. 2020, 18, 39.
- Gangat, A.A.; Te, I.; Kao, Y.-J. Steady States of Infinite-Size Dissipative Quantum Chains via Imaginary Time Evolution. Phys. Rev. Lett. 2017, 119, 010501.
- Kageyama, S.; Ikeda, H.; Miyahara, Y.; Imai, N.; Ishihara, M.; Saito, K.; Sugino, S.; Ueda, S.; Ishikawa, T.; Kokura, S.; et al. Adoptive Transfer of MAGE-A4 T-cell Receptor Gene-Transduced Lymphocytes in Patients with Recurrent Esophageal Cancer. Clin. Cancer Res. 2015, 21, 2268–2277.
- Jackson, H.J.; Rafiq, S.; Brentjens, R.J. Driving CAR T-cells forward. Nat. Rev. Clin. Oncol. 2016, 13, 370–383.
- Neelapu, S.S.; Locke, F.L.; Bartlett, N.L.; Lekakis, L.J.; Miklos, D.B.; Jacobson, C.A.; Braunschweig, I.; Oluwole, O.O.; Siddiqi, T.; Lin, Y.; et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma. N. Engl. J. Med. 2017, 377, 2531–2544.
- Bejarano, L.; Bosso, G.; Louzame, J.; Serrano, R.; Gómez-Casero, E.; Martinez-Torrecuadrada, J.L.; Martínez, S.; Blanco-Aparicio, C.; Pastor, J.; Blasco, M.A. Multiple cancer pathways regulate telomere protection. EMBO Mol. Med. 2019, 11, 10292.
- Mu, X.; Sang, Y.; Fang, C.; Shao, B.; Yang, L.; Yao, K.; Zhao, X.; Gou, J.; Wei, Y.; Yi, T.; et al. Immunotherapy of tumors with human telomerase reverse transcriptase immortalized human umbilical vein endothelial cells. Int. J. Oncol. 2015, 47, 1901–1911.
- Cesare, A.; Reddel, R. Alternative lengthening of telomeres: Models, mechanisms and implications. Nat. Rev. Genet. 2010, 11, 319–330.
- Slatter, T.L.; Tan, X.; Yuen, Y.C.; Gunningham, S.; Ma, S.S.; Daly, E.; Packer, S.; Devenish, C.; Royds, J.A.; Hung, N.A. The alternative lengthening of telomeres pathway may operate in non-neoplastic human cells. J. Pathol. 2011, 226, 509–518.
- Dilley, R.L.; Greenberg, R.A. ALTernative Telomere Maintenance and Cancer. Trends Cancer 2015, 1, 145–156.
- Fan, H.-C.; Chen, C.-M.; Chi, C.-S.; Tsai, J.-D.; Chiang, K.-L.; Chang, Y.-K.; Lin, S.-Z.; Harn, H.-J. Targeting Telomerase and ATRX/DAXX Inducing Tumor Senescence and Apoptosis in the Malignant Glioma. Int. J. Mol. Sci. 2019, 20, 200.
- Sommer, A.; Royle, N.J. ALT: A Multi-Faceted Phenomenon. Genes 2020, 11, 133.
- Dyer, M.A.; Qadeer, Z.; Valle-Garcia, D.; Bernstein, E. ATRX and DAXX: Mechanisms and Mutations. Cold Spring Harb. Perspect. Med. 2017, 7, a026567.
- Yang, X.; Khosravi-Far, R.; Chang, H.Y.; Baltimore, D. Daxx, a Novel Fas-Binding Protein That Activates JNK and Apoptosis. Cell 1997, 89, 1067–1076.
- Gibbons, R.J.; McDowell, T.L.; Raman, S.; O’Rourke, D.M.; Garrick, D.; Ayyub, H.; Higgs, D.R. Mutations in ATRX, encoding a SWI/SNF-like protein, cause diverse changes in the pattern of DNA methylation. Nat. Genet. 2000, 24, 368–371.
- Gibbons, R. Alpha thalassaemia-mental retardation, X linked. Orphanet J. Rare Dis. 2006, 1, 15.
- Brosnan-Cashman, J.A.; Yuan, M.; Graham, M.K.; Rizzo, A.J.; Myers, K.M.; Davis, C.; Zhang, R.; Esopi, D.M.; Raabe, E.H.; Eberhart, C.G.; et al. ATRX loss induces multiple hallmarks of the alternative lengthening of telomeres (ALT) phenotype in human glioma cell lines in a cell line-specific manner. PLoS ONE 2018, 13, e0204159.
- Ro, C.; Chai, W.; Yu, V.E.; Yu, R. Pancreatic neuroendocrine tumors: Biology, diagnosis, and treatment. Chin. J. Cancer 2013, 32, 312–324.
- Jiao, Y.; Shi, C.; Edil, B.H.; de Wilde, R.F.; Klimstra, D.S.; Maitra, A.; Schulick, R.D.; Tang, L.H.; Wolfgang, C.L.; Choti, M.A.; et al. DAXX/ATRX, MEN1, and mTOR Pathway Genes Are Frequently Altered in Pancreatic Neuroendocrine Tumors. Science 2011, 331, 1199–1203.
- Heaphy, C.M.; de Wilde, R.F.; Jiao, Y.; Klein, A.P.; Edil, B.H.; Shi, C.; Bettegowda, C.; Rodriguez, F.J.; Eberhart, C.G.; Hebbar, S.; et al. Altered Telomeres in Tumors with ATRX and DAXX Mutations. Science 2011, 333, 425.
- Pekmezci, M.; Rice, T.; Molinaro, A.M.; Walsh, K.; Decker, P.A.; Hansen, H.; Sicotte, H.; Kollmeyer, T.M.; McCoy, L.S.; Sarkar, G.; et al. Adult infiltrating gliomas with WHO 2016 integrated diagnosis: Additional prognostic roles of ATRX and TERT. Acta Neuropathol. 2017, 133, 1001–1016.
- Clynes, D.; Higgs, D.; Gibbons, R. The chromatin remodeller ATRX: A repeat offender in human disease. Trends Biochem. Sci. 2013, 38, 461–466.
- Napier, C.E.; Huschtscha, L.I.; Harvey, A.; Bower, K.; Noble, J.R.; Hendrickson, E.A.; Reddel, R.R. ATRX represses alternative lengthening of telomeres. Oncotarget 2015, 6, 16543–16558.
- Killela, P.J.; Reitman, Z.J.; Jiao, Y.; Bettegowda, C.; Agrawal, N.; Diaz, L.A., Jr.; Friedman, A.H.; Friedman, H.; Gallia, G.L.; Giovanella, B.C.; et al. TERT promoter mutations occur frequently in gliomas and a subset of tumors derived from cells with low rates of self-renewal. Proc. Natl. Acad. Sci. USA 2013, 110, 6021–6026.
- Flynn, R.L.; Cox, K.E.; Jeitany, M.; Wakimoto, H.; Bryll, A.R.; Ganem, N.J.; Bersani, F.; Pineda, J.R.; Suvà, M.L.; Benes, C.H.; et al. Alternative lengthening of telomeres renders cancer cells hypersensitive to ATR inhibitors. Science 2015, 347, 273–277.
- Shay, J.W.; Reddel, R.R.; Wright, W.E. Cancer and Telomeres—An ALTernative to Telomerase. Science 2012, 336, 1388–1390.
- Kosiol, N.; Juranek, S.; Brossart, P.; Heine, A.; Paeschke, K. G-quadruplexes: A promising target for cancer therapy. Mol. Cancer 2021, 20, 1–18.
- Chhabra, G.; Wojdyla, L.; Frakes, M.; Schrank, Z.; Leviskas, B.; Ivancich, M.; Vinay, P.; Ganapathy, R.; Ramirez, B.E.; Puri, N. Mechanism of Action of G-Quadruplex–Forming Oligonucleotide Homologous to the Telomere Overhang in Melanoma. J. Investig. Dermatol. 2018, 138, 903–910.
- Mender, I.; Gryaznov, S.; Dikmen, Z.G.; Wright, W.E.; Shay, J.W. Induction of telomere dysfunction mediated by the telomerase substrate precursor 6-thio-2′-deoxyguanosine. Cancer Discov. 2014, 5, 82–95.
- Naderlinger, E.; Holzmann, K. Epigenetic Regulation of Telomere Maintenance for Therapeutic Interventions in Gliomas. Genes 2017, 8, 145.
- Dogan, F.; Forsyth, N. Telomerase Regulation: A Role for Epigenetics. Cancers 2021, 13, 1213.