2. Sphingolipid Synthesis, Catabolism, and Trafficking in Yeast
In
S. cerevisiae, the major pathways and enzymes involved in sphingolipid synthesis have already been identified (
Figure 1A). Sphingolipid biosynthesis begins with the condensation of cytosolic serine and palmitoyl-CoA by the ER-resident serine palmitoyltransferase (SPT) complex, yielding 3-ketodihydrosphingosine (KDS). The SPT complex contains three subunits: Lcb1, Lcb2, and Tsc3. Because its active site is at the interface of the two catalytic subunits, Lcb1 and Lcb2, facing the cytosol, it is suspected that KDS is incorporated into the outer leaflet of the ER membrane after its synthesis [
13,
14,
15]. Subsequently, KDS is reduced to dihydrosphingosine (DHS) or, in a following step, hydroxylated to phytosphingosine (PHS), by enzymes that also face the outer leaflet of the ER [
16]. DHS and PHS are termed “LCBs,”, which act as the backbone of sphingolipids. Both LCB-types can be converted with a CoA-activated very long-chain fatty acid (VLCFA) into dihydroceramide and phytoceramide, respectively, through ceramide synthase. Ceramide synthase is a heteromeric protein complex, containing three subunits, Lag1, Lac1, and Lip1. Lag1 and Lac1 are homologous proteins that feature eight transmembrane domains (TMDs) and a conserved Lag motif, which extends over the 5th and 6th TMDs and presumably harbor the active site of the proteins [
17]. Both proteins are functionally similar, but differ in their substrate specificity, with Lag1 being better than Lac1 in converting PHS to phytoceramide [
18]. This difference already accounts for the distinct roles of Lag1 and Lac1 in aging and further suggests that each LCB isoform and its derivatives have unique physiological functions [
18]. The third subunit of the ceramide synthase Lip1 features a single TMD and exerts its essential function in the complex through the transmembrane or luminal part of the protein [
19]. The topology of ceramide synthase is not understood in detail. However, a model was proposed, in which the catalytic center of the ceramide synthase forms a channel that connects the leaflets of the ER membrane [
17]. Translocation between the ER leaflets is essential for both the substrate and product of ceramide synthase. For example, after a series of phosphorylation and dephosphorylation steps, exogenously added DHS is delivered into the inner layer of the ER, where it is used for ceramide synthesis [
20,
21]. Therefore, translocation is necessary to ensure the conversion of endogenously synthesized and exogenously added DHS. Likewise, ceramide is found on both sides of the ER membrane and can be transported by vesicular transport from either side to the next destination of the sphingolipid synthesis pathway, the Golgi. Ceramides present on the cytosolic side are also available for delivery to the Golgi via non-vesicular transport.
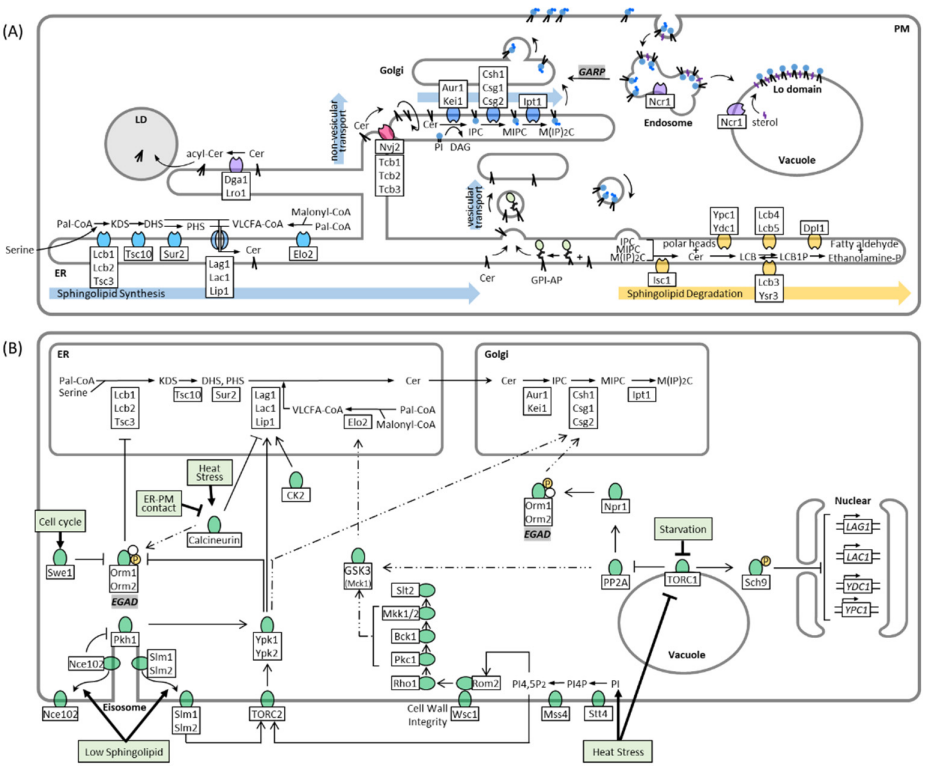
The transport of ceramide from the ER to the Golgi depends mainly on the COPII vesicles of the secretory pathway, similar to that of proteins. Glycosylphosphatidylinositol (GPI)-anchored proteins are also synthesized in the ER and transported to the Golgi apparatus by COPII vesicles [
24]. Ceramides in the inner layer of the ER are involved in remodeling GPI lipid moieties and seem to coalesce with GPI-anchored proteins during vesicular transport (
Figure 1A), as defects in GPI-anchored biosynthesis affect vesicular transport of ceramide [
25,
26,
27]. Conversely, normal ceramide synthesis is required for GPI-anchored protein transport [
28]. GPI-anchored proteins are segregated into lipid microdomains in the ER and are sorted into specific transport vesicles [
29]. Recent observations, using high-speed, super-resolution live imaging microscopy, have confirmed this model and suggest that the length of ceramide in the raft-like structure is important for the sorting mechanism [
30]. Although vesicular transport is the main pathway for ceramide to reach the Golgi, alternative routes exist that maintain some transport even under conditions that block the secretory pathway [
31]. This so-called non-vesicular ceramide transport depends on tethering proteins at ER-Golgi contact sites, which facilitate the transfer of ceramide between the surface of the ER and Golgi. The three members of the tricalbin protein family Tcb1, Tcb2, and Tcb3, together with Nvj2 were proposed as inducible tethering proteins that re-localize during ER stress from their constitutive sites to ER-Golgi contact sites to facilitate non-vesicular ceramide transport [
12,
113]. In addition, a study with a reconstituted cell-free system showed that non-vesicular ceramide transport involves heat-sensitive cytosolic protein(s) [
31]. In mammalian cells, CERT is a well-described soluble transfer protein that delivers ceramide to the Golgi at ER-Golgi contact sites [
118,
119].
Ceramide is converted to complex sphingolipids in the Golgi apparatus. In
S. cerevisiae, the major complex sphingolipid is inositol phosphorylceramide (IPC), which is synthesized by IPC synthase via the transfer of an inositol phosphate headgroup from phosphatidylinositol to ceramide. IPC synthase is a protein complex with two known membrane-spanning subunits, Aur1 and Kei1. While Aur1 contains the active site of the complex that faces the lumen of the Golgi [
32], Kei1 was shown to be essential for the localization of the complex and its catalytic ability [
33]. Because ceramides transported by the non-vesicular pathway are incorporated into the outer layer of the Golgi and used for IPC synthesis [
31], it is apparent that a yet unidentified mechanism translocates ceramides to the luminal site. After its synthesis, IPC can be further modified by the mannosylation of its headgroup to form mannose inositolphosphorylceramide (MIPC) or mannose di (inositolphosphoryl) ceramide (M(IP)
2C). The synthesis of MIPC depends on the Csh1p/Csg1p/Csg2p complex, and the active site of Csg1p is also predicted in the lumen of the Golgi [
34]. Notably, the catalytic subunits of the complex show a different affinity to the dihydro- and phyto-isoforms of IPC, such as the previously mentioned ceramide synthase subunits [
35]. Conversion of MIPC to M(IP)
2C is carried out by Ipt1 [
35,
36], and complex sphingolipids are transported to the PM. This transport is once more coupled to vesicular transport, as it was shown that the transport of IPC, MIPC, and M(IP)
2C to the PM almost completely stops when vesicular transport is blocked [
22,
37].
The remodeling of membranes, in response to cell cycle progression and environmental changes, requires enzymes that can reverse each step of the sphingolipid synthesis pathway. Inositol-phosphosphingolipid phospholipase C (Isc1) localizes to the ER and mitochondrial outer membrane and cleaves the polar head group from complex sphingolipids at the C1 position, yielding phytoceramide and a free head group [
38,
39] (
Figure 1A and
Figure 2C). ER and endosomal enzymes degrade phytoceramide in multiple steps into PHS and 2-hydroxyhexadecanal, which can be recycled into glycerophospholipid metabolism [
40,
41,
42]. Defects in sphingolipid degradation and recycling are connected to a variety of human lysosomal storage diseases such as Niemann Pick type C, a fatal neurodegenerative disorder caused by mutations in the
NPC1 or
NPC2 gene, which is characterized by lysosomal accumulation of cholesterol and sphingolipids [
43]. In yeast, sphingolipid breakdown is also connected to endosomal and vacuolar degradation because deletion of Isc1 leads to defective lysosomal trafficking and vacuolar function [
44,
45]. Cells lacking Isc1 not only build up complex sphingolipids but also some sphingolipid precursors, including very long-chain ceramide species that are presumably derived from increased de novo synthesis [
46]. Increased levels of long-chain ceramides activate the catalytic subunit of the protein phosphatase type 2A (PP2A) complex Sit4, which links ceramide and vacuolar homeostasis by negatively regulating protein sorting, vesicular trafficking, vacuole function, and autophagy [
44]. Consequently, double deletion of Sit4 and Isc1 restores vacuole function [
44]. Sphingolipids not only control endosomal maturation via Sit4 but are also sorted and recycled themselves on endosomes. Ncr1/2, yeast orthologs to the human Npc1/2 proteins, were suggested to sort sphingolipids on endosomes with IPC being directed to the vacuolar membrane, and MIPC and M(IP)2C being returned to the Golgi and subsequently transported to the PM [
47]. The recycling of PM sphingolipids depends on the Golgi-associated retrograde protein (GARP) complex, which tethers retrograde transport vesicles derived from endosomes to the late Golgi [
48]. Blocking this pathway induces toxic accumulation of LCBs, which suggests increased degradation of complex sphingolipids in the vacuole [
49]. In addition to sphingolipid sorting, Ncr1/2 plays a role in sterol transport from endosomes to the vacuolar membrane to form sterol-and sphingolipid-enriched raft-like domains that mediate microautophagy [
50]. Because GARP deficiency also alters sterol metabolism [
49], it seems likely that sphingolipids and sterols are sorted via the same Ncr1/2-mediated mechanism.
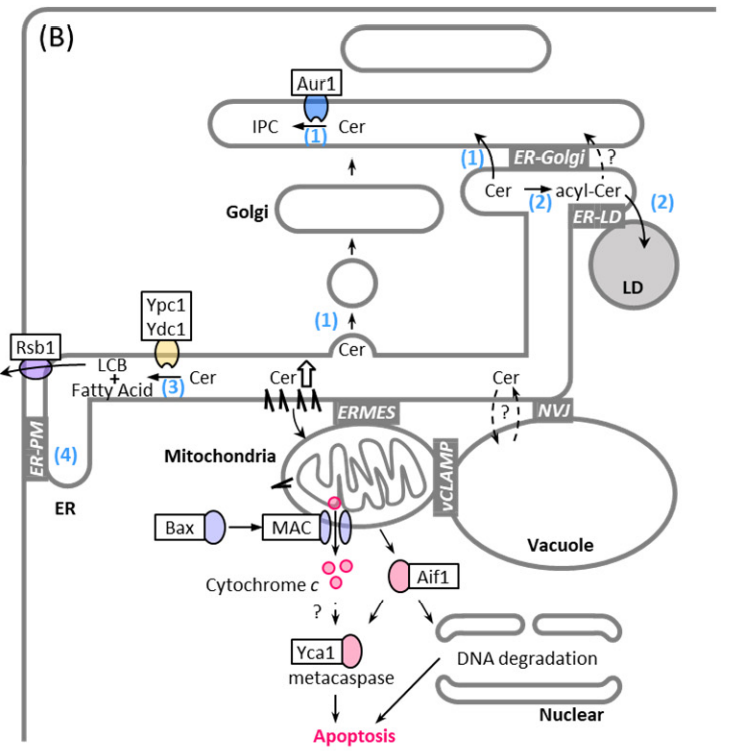
Figure 2. Mechanical scheme of ceramide-mediated apoptosis and pathways of ceramide removal from the ER: ceramide transfer to the Golgi and conversion into IPC (1), ceramide conversion into acylceramide and storage in the LDs (2), ceramide hydrolyzation into LCB and free fatty acid (3), and LCB transport from the cytoplasmic side toward the extracytoplasmic side [
120], probably at ER-PM contact site (4). Abbreviations: mitochondrial apoptosis-induced channel (MAC), nucleus-vacuole junction (NVJ), vacuole and mitochondria patch (vCLAMP), endoplasmic reticulum-mitochondria encounter structure (ERMES).
3. Regulation of Sphingolipid Metabolism
LCBs, ceramides, and other intermediates in sphingolipid metabolism are vital but potentially toxic signaling molecules [
2,
3]. In
S. cerevisiae, inhibition of IPC synthase activity by aureobasidin A, a specific inhibitor of Aur1, leads to cell death or growth inhibition [
121,
122,
123,
124,
125,
126,
127]. This is thought to be due to the reduction of complex sphingolipids or accumulation of ceramides, which leak into mitochondria by a not understood mechanism and induce the mitochondrial apoptotic pathway [
123,
124,
127,
128,
129] (
Figure 2). Supplying complex sphingolipids to the PM based on need and simultaneously preventing the toxic accumulation of metabolites is an intricate task for cells. It is, therefore, not surprising that many partially interconnected pathways exist that respond to environmental changes and regulate different steps along the sphingolipid synthesis pathway (
Figure 1B). The formation of LCBs from serine and palmitoyl-CoA by SPT is the rate-limiting step of sphingolipid synthesis and is the main target of regulation. It was shown that LCB synthesis depends on the external uptake of serine, indicating that sphingolipid metabolism is not regulated prior by de novo serine production [
51]. The catalytic activity of the SPT complex mainly depends on the presence of two regulatory proteins, Orm1 and Orm2, which inhibit SPT when they are associated with it [
52,
53]. Orm1/2 are downstream effector proteins of an extensive kinase network that coordinates sphingolipid metabolism with other cellular tasks, such as cell cycle progression, nutrient uptake, and stress response. Here, we break down the pathways of this kinase network targeting Orm1/2 starting with the initial stimuli:
Sphingolipid deficiency: PM stress caused by sphingolipid deficiency is accompanied by changes in membrane properties and rearrangement of lateral microdomains. Slm1, Slm2, and Nce102 are proteins that localize at steady state to the eisosome, a protein complex that is part of the static PM microdomain, known as the membrane compartment which is occupied by Can1 (MCC) (
Figure 1B). Slm1/2 are proteins that localize to the PM by binding to phosphatidylinositol 4,5-bisphosphate (PI4,5P2), whereas Nce102 is an integral PM protein. Depletion of sphingolipids disrupts the lipid and protein organization of the MCC, causing Slm1/2 and Nce102 to dissociate from the eisosome within the membrane plane. In this way, Slm1/2 become available to bind and activate the PM resident TORC2, which in turn phosphorylates and activates the cytosolic protein kinase Ypk1 [
7,
54]. Similar to Slm1/2, dissociation of Nce102 alleviates its inhibitory effect on the eisosome resident protein kinases Pkh1 and Pkh2, which in turn phosphorylate Ypk1 as well, but at different sites compared to TORC2-mediated phosphorylation [
55]. Ypk1 phosphorylates several targets, including Orm1/2, triggering their release from the SPT complex and stopping SPT inhibition [
7,
53,
56]. Phosphorylation of Orm1/2 not only causes the proteins to dissociate from the SPT complex but leads to their degradation, via the recently identified endosome and Golgi-associated degradation (EGAD) pathway [
57]. In this pathway, phosphorylated Orm2 is exported to the Golgi and endosomes, where it is selectively polyubiquitinated by the Dsc ubiquitin ligase complex, extracted from the membrane by the unfoldase Cdc48, and finally degraded by the proteasome [
57]. Degradation of already inactivated Orm2 is necessary because accumulation of Orm2 can divert Ypk1 activity away from other important targets, such as ceramide synthase [
58]. Unlike the SPT complex, which depends on Orm1/2 interaction, ceramide synthase and many other key enzymes in sphingolipid biosynthesis are regulated by direct phosphorylation through protein kinases. Both the ceramide synthase subunits Lag1 and Lac1 are phosphorylated by Ypk1 to activate ceramide synthesis [
58,
59]. Consequently, TORC2/Ypk1 action controls the first two consecutive steps of sphingolipid synthesis, which ensures a coherent flux of metabolites [
58] and provides a feedback mechanism, in response to sphingolipid depletion [
7]. Moreover, Ypk1 was suggested to further regulate complex sphingolipid synthases such as Csh1 and Sur1 as these enzymes are phosphorylated at Ypk1 typical phosphorylation motifs, in response to myriocin-induced SPT inhibition [
60].
Cell cycle progression and growth: The cell cycle checkpoint kinase Swe1 induces Orm1/2 phosphorylation and LCB synthesis independently of Ypk1 [
61]. Swe1 is activated and halts cell cycle progression in response to defective lipolysis, which is a source of lipid precursors for sphingolipid synthesis [
62]. Therefore, Orm1/2 phosphorylation by Swe1 could be a feedback loop to maintain a steady supply of LCBs. Moreover, casein kinase 2 (CK2), a major regulator of various pro-growth cellular events and suppressor of apoptosis [
63,
64], promotes ceramide synthesis by directly phosphorylating the
C-terminal cytoplasmic domains of the ceramide synthase subunits Lag1 and Lac1 [
65]. CK2 dependent phosphorylation of ceramide synthase was suggested to maintain its localization to the ER membrane via the COP I-dependent C-terminal dilysine ER retrieval pathway [
65].
Starvation: Orm1/2 are also phosphorylated via the TORC1 pathway (
Figure 1B), which senses amino acids at the vacuole and adjusts cellular metabolism in response to amino acid starvation [
66]. TORC1 is inhibited under these conditions and is unable to phosphorylate a variety of downstream targets, including the cytosolic protein phosphatase type 2A (PP2A), which is suppressed by TORC1 at steady state [
67]. PP2A is responsible for dephosphorylation and activation of the protein kinase Npr1, which in turn phosphorylates Orm1/2 but at different sites compared to Ypk1 [
6]. In contrast, Npr1 mediated Orm1/2 phosphorylation does not affect LCB synthesis, but instead activates Orm1/2 to promote the synthesis of complex sphingolipids downstream of the SPT complex through an unidentified mechanism [
6]. Because increased complex sphingolipid levels at the PM facilitate the integration of amino acid permeases, TORC1 inhibition provides a feedback loop to promote nutrient uptake during starvation [
6]. Nevertheless, it was shown that TORC1 controls the early steps of sphingolipid metabolism by mechanisms other than Orm1/2 phosphorylation. The introduced PP2A branch of TORC1 was proposed to control the phosphorylation and activity of the VLCFA elongase Elo2 via Mck1 kinase [
68]. The details of this pathway remain unclear, and contradicting reports have been published on whether Mck1 mediated phosphorylation of Elo2 promotes [
68] or inhibits Elo2 activity [
69]. Furthermore, steady-state levels of sphingolipids are regulated through the Sch9 effector branch of TORC1, which represses the expression of the ceramidase genes YDC1 and YPC1 as well as ceramide synthase genes LAG1 and LAC1 [
70]. Conversely, Sch9 seems to mediate hydrolytic ceramide production during diauxic shift, as Sch9 is essential for the proper translocation of inositol phosphospingolipid phospholipase C, Isc1, from the ER to the mitochondria [
70]. Whereas the main role of TORC1 is to adjust cellular metabolism to amino acid availability, other stressors regulate TORC1 function as well such as carbon or phosphorous starvation, hyperosmolarity, oxidants and heat [
71,
72].
Heat stress and cell wall integrity (CWI) defects: Heat stress induces TORC1 sequestration into stress granules, thereby inhibiting TORC1 function in the vacuole and activating sphingolipid synthesis in a way that is similar to starvation [
73]. In addition, elevated temperature is another cause of PM stress that induces sphingolipid synthesis by promoting phosphoinositide-signaling at the PM and subsequently activates two parallel responses, the previously introduced TORC2 pathway and the CWI pathway. Phosphatidylinositides give organellar membranes characteristic properties and drive many membrane-related processes by recruiting effector proteins that specifically recognize their phosphorylation patterns [
74]. The two characteristic phosphoinositides of the PM are phosphatidylinositol 4-phosphate (PI4P) and PI(4,5)P2, which are synthesized by the subsequent kinase activities of Stt4 and Mss4, respectively. PM phosphoinositides and sphingolipids seem to be mutually regulated because the localization and activity of Mss4 depends on sphingolipid synthesis [
75,
76]. Conversely, it was shown that the substrate of Mss4, PI4P, accumulates at the PM upon inhibition of sphingolipid synthesis [
77]. Heat shock induces a transient increase in PM phosphoinositides, which recruit PI(4,5)P2-binding Slm1/2 proteins as well as Pkh1 to the PM [
78,
79] and promote the Pkh-dependent phosphorylation of Slm1/2 [
80]. Thus, sphingolipid synthesis is activated via the Pkh1-TORC2-Ypk1 pathway. Simultaneously, elevated levels of phosphoinositides recruit the guanine nucleotide exchange factors Rom1 and 2 to the PM [
79], which together with cell-surface sensors Wsc1, 2 and 3 stimulate nucleotide exchange on the small G protein, Rho1, the designated master regulator of the CWI pathway [
81]. The CWI pathway controls several stress response mechanisms, including the high osmolarity response pathway, which alleviates the accumulation of aberrant reactive oxygen species caused by heat stress or inhibited sphingolipid synthesis [
82,
83,
84]. In contrast, it was shown that the VLCFA elongase Elo2 is phosphorylated and inactivated by Mck1 kinase, a presumed downstream effector of the CWI pathway (
Figure 1B), and a model was proposed in which CWI signaling is inactivated in response to perturbed sphingolipid synthesis to de-repress Elo2 [
69]. However, this model requires additional investigation, as it remains unclear how signals from the PM are transduced to Elo2 and how Elo2 inhibition is coordinated with other CWI functions.
Heat induced sphingolipid synthesis last approximately 30 min after which LCBs reach near-basal levels [
36,
85]. Timely monitoring of sphingolipid synthesis and Orm2 phosphorylation in response to heat stress revealed that Orm2 phosphorylation decreases when LCBs reach peak levels, while ceramide synthesis peaks 10 min later [
86]. As this effect is completely diminished by inhibition of SPT and accelerated by exogenously adding LCBs it was proposed that LCBs activate protein phosphatases that act upstream of Orm1/2 to restore SPT inhibition after the heat response. The protein phosphatase PP2A, which is also activated by ceramides [
44], has been proposed to mediate inhibition of Ypk1 [
86] or Pkh1/2 [
87].
The calcium activated protein phosphatase calcineurin acts as a major antagonist to the TORC2 pathway and reverses Lag1 and Lac1 phosphorylation [
58,
59], activates ORM2 transcription [
8] and represses Orm1/2 degradation [
101]. Studies proposed that ER-PM contact sites, particularly the cortical ER-resident tricalbin proteins, could contain calcineurin activity and finetune the TORC2/calcineurin rheostat.[
90,
100,
106,
107].
Besides regulation of sphingolipid synthesis through the kinase network, other mechanism are involved in preventing toxic ceramide accumulation. Ceramide conversion into acylceramide by the ER and lipid droplet (LD)-resident enzymes Dga1, and Lro1 eneables acylceramide uptake into LDs and seems to prevent toxic ceramide leakge into mitochondria [
10,
11,
12]. Because the deletion of Nvj2, Dga1, and Lro1 leads to a synthetic growth defect [
113], ER-Golgi contact-mediated ceramide transport and acylceramide synthesis seems to be redundant in the clearance of toxic ceramides. Combinational mutations of tricalbins and Sec proteins that drive vesicular transport cause a dramatic accumulation of LDs [
12], suggesting that vesicular transport from the ER is also important in preventing ceramide stress (
Figure 2). Given that excess ceramides are converted into acylceramides in the ER and acylceramides are incorporated into the LDs [
10,
11,
12,
113], ER-LD contacts may play a role in protection against ceramide toxicity. Consistent with this, a recent study showed that loss of ER-LD contacts results in increased levels of ceramides and their sphingoid precursors [
130], even though its effect on acylceramide levels remains unclear. Interestingly, it was suggested that ER-LD contacts are one of the places where sphingoid intermediates are synthesized, and that seipin, an ER protein involved in LD biogenesis, negatively regulates sphingoid production at ER-LD contact sites [
130] (
Figure 1A). Thus, ER-LD contact sites may help prevent the accumulation of toxic ceramide not only by facilitating the formation of acylceramide and its incorporation into the LD, but also by reducing the levels of sphingoid intermediates.