The active form of vitamin D, 1α,25-(OH)2D3, not only promotes intestinal calcium absorption, but also regulates the formation of osteoclasts (OCs) and their capacity for bone mineral dissolution. Gal-3 is a newly discovered bone metabolic regulator involved in the proliferation, differentiation, and apoptosis of various cells.
- galectin-3
- 1α
- 25-(OH)2D3
- osteoclasts
- bone resorption
1. Introduction
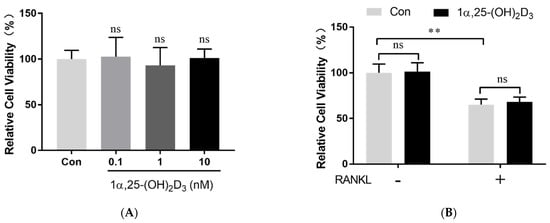
2. 1α,25-(OH)2D3 Had No Effect on Osteoclast Precursor Viability
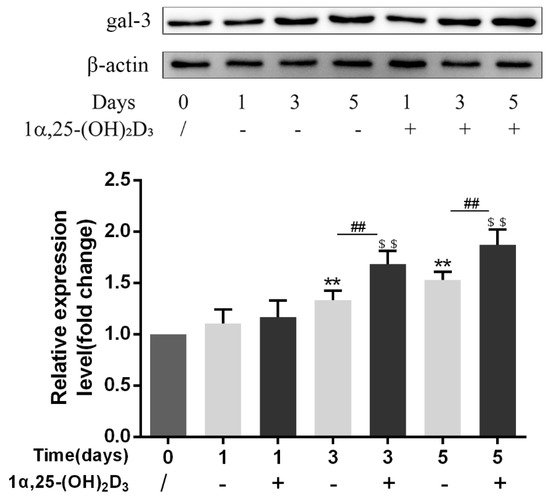
3. 1α,25-(OH)2D3 Promoted Gal-3 Expression

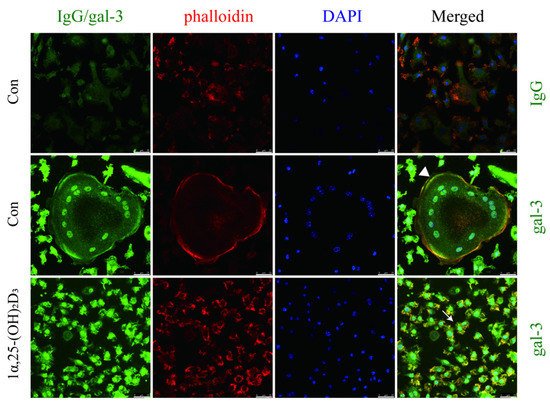
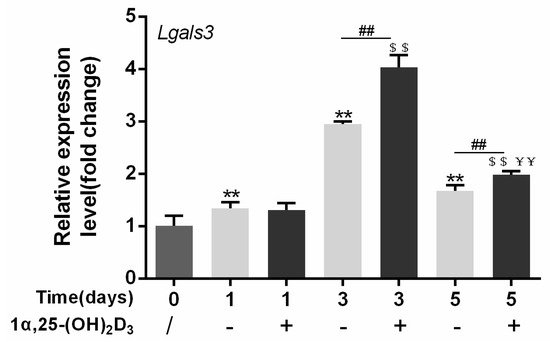
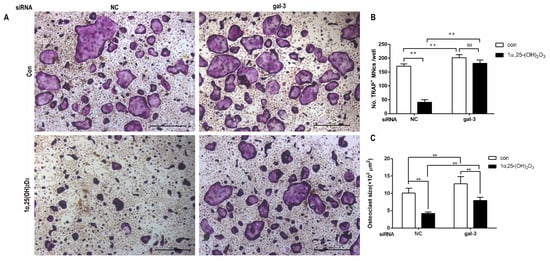
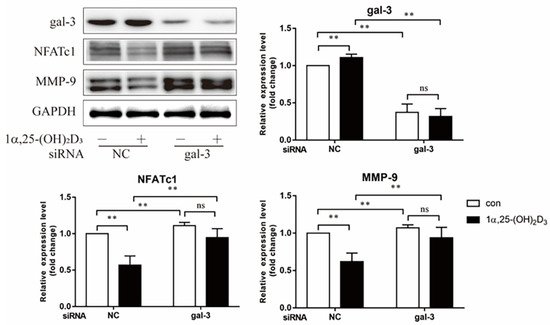
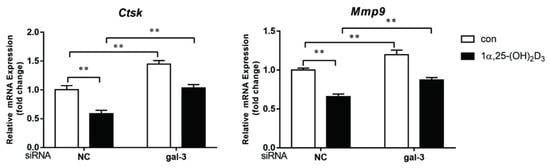
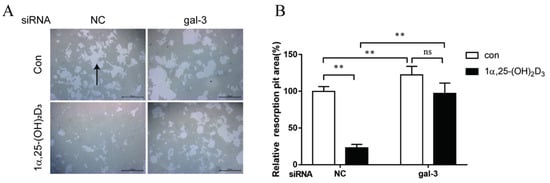
4. Gal-3 Contributed to Osteoclasts Formation and Activation Regulated by 1α,25-(OH)2D3
5. Interaction between Gal-3 and VDR
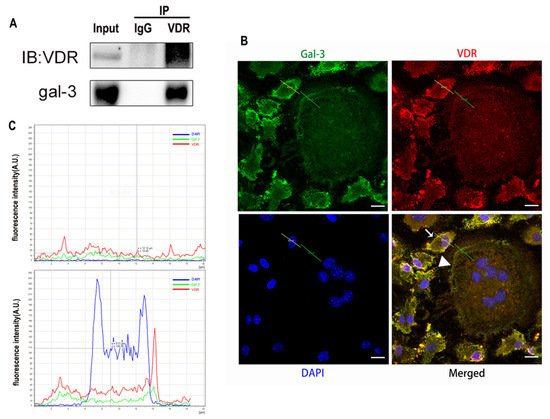
3. Discussion
Vitamin D is known as the anti-rickets vitamin, and the active form 1α,25-(OH)2D3 is involved in a variety of physiological processes. Among these physiological processes, the most important function is the regulation of calcium homeostasis together with PTH. When serum calcium is low, 1α,25-(OH)2D3 acts with PTH to increase calcium absorption from the intestine. If normal calcium cannot be maintained by intestinal calcium absorption, 1α,25-(OH)2D3 will act with PTH to increase the reabsorption of calcium from the kidney and increase calcium release from the bone stores [32]. Kitazawa et al. [33] also reported that 1α,25-(OH)2D3 increases calcium release from bone stores. They confirmed that 1α,25-(OH)2D3 can increase RANKL expression in stromal cells and support osteoclastogenesis indirectly. In addition, 1α,25-(OH)2D3 can also act directly on OCPs and conversely inhibit OC formation. Consistent with our previous results [34], we demonstrated that 1α,25-(OH)2D3 directly inhibited TRAP-positive OC formation. We also confirmed that 1α,25-(OH)2D3 inhibited the expression of OC-related genes and proteins, including Ctsk, MMP-9, and NFATc1 expression. The bone resorption activity of OCs was also inhibited by 1α,25-(OH)2D3. These data are consistent with those previous studies. For example, Sakai et al. [35] also found that 1α,25-(OH)2D3 inhibits OC formation by inhibiting the expression of c-Fos and NFATc1. Kikuta et al. [36] confirmed that 1α,25-(OH)2D3 and its analogue eldecalcitol promoted OPCs migration and inhibited OC formation by regulating the sphingosine1-phosphate (S1P) receptor system. Thus, we hypothesize that 1α,25-(OH)2D3 can both promote and inhibit OC formation under different circumstances to achieve bone health equilibrium based on previous research. Therefore, the effect of 1α,25-(OH)2D3 on bone health and its underlying mechanism should be further elucidated. In a previous study, we found that gal-3 expression changes during OC formation in the presence of 1α,25-(OH)2D3 by chance [23]. Here, we further confirmed that 1α,25-(OH)2D3 increases gal-3 expression at the protein and mRNA levels. We then used gal-3-knockdown OCPs to evaluate OC formation, bone resorption, and the expression of OC-related genes and proteins.
Gal-3 is the only member of chimera-type galectins and is structurally composed of a large N-terminal domain and one carbohydrate recognition domain (CRD). It contains three structurally distinct domains: (1) a short amino terminal consisting of 12 amino acids with a serine phosphorylation site responsible for its translocation [37]; (2) a collagen-alpha-like nearly 110 amino acid long structure rich in proline, alanine, and glycine; and (3) a C-terminal domain with nearly 140 amino acids encompassing the CRD [38]. It participates in a variety of cellular processes, such as proliferation, differentiation, migration, and apoptosis, and has a variety of biological effects, especially in nephropathy, carcinoma, and the quiescence of hematopoietic stem cells [39][40][41]. It is also an important regulator of bone remodeling. Iacobini et al. [42] found that Lgals3−/− OBs and OCs showed impaired terminal differentiation, reduced mineralization capacity, and resorption activity. They confirmed that gal-3 is an essential factor in normal osteocyte differentiation and activity, bone reconstruction, and biomechanical balance. Simon et al. [24] proved that extracellular gal-3 inhibited OC formation and gal-3-deficient bone marrow cells displayed a higher osteoclastogenic capacity. In the present study, we also found that gal-3-knockdown OCPs showed a stronger OC formation capacity and bone resorption capacity than those of normal OCPs. The expression levels of OC-related genes and proteins, including CTSK, MMP-9, and NFATc1, increased after gal-3 knockdown. As reported previously [24], our data also suggested that gal-3 is a negative regulator of OC formation and activation. In this study, we also found that 1α,25-(OH)2D3 not only inhibited OC formation and activation but also increased gal-3 expression in mRNA and protein levels. However, the mRNA expression of gal-3 in day 5 is lower than that in day 3. This might be relative to mRNA translation and characteristic of protein expression. It has been reported that some of the gal-3 crossed the membrane and translocated into the intracellular vesicles and/or directly onto the cell surface. Furthermore, cell surface-bound and extracellular gal-3 may re-enter the cell by endocytosis and take part in a recycling loop, and is then found in the vesicular non-cytosolic compartments [43]. This may be the cause of the inconsistent expression of gal-3 protein and mRNA, which needs further investigation. When gal-3 expression was inhibited by small interfering RNAs, the inhibitory effect of 1α,25-(OH)2D3 on OC formation and activation was significantly alleviated. These data suggest that gal-3 contributes to 1α,25-(OH)2D3-mediated OC formation and activity. Immunoprecipitation and immunofluorescence assays supported the interaction between gal-3 and VDR protein, further demonstrating that gal-3 might participate in OC formation and activation via interaction with VDR induced by 1α,25-(OH)2D3. However, Nakajima et al. [22] suggested that intact gal-3 promotes OC formation, whereas cleaved gal-3 inhibits OC formation. Therefore, the role of gal-3 in the regulation of OC formation and activation by vitamin D remains to be elucidated. In any case, gal-3 is a potential new target for the prevention of bone disease and maintenance of calcium homeostasis.This entry is adapted from the peer-reviewed paper 10.3390/ijms222413334
References
- William J. Boyle; W. Scott Simonet; David L. Lacey; Osteoclast differentiation and activation. Nature 2003, 423, 337-342, 10.1038/nature01658.
- Megan M. Weivoda; Chee Kian Chew; David G. Monroe; Joshua N. Farr; Elizabeth J. Atkinson; Jennifer R. Geske; Brittany Eckhardt; Brianne Thicke; Ming Ruan; Amanda J. Tweed; et al. Identification of osteoclast-osteoblast coupling factors in humans reveals links between bone and energy metabolism. Nature Communications 2020, 11, 1-13, 10.1038/s41467-019-14003-6.
- Brendan F. Boyce; Advances in osteoclast biology reveal potential new drug targets and new roles for osteoclasts. Journal of Bone and Mineral Research 2013, 28, 711-722, 10.1002/jbmr.1885.
- Anaïs Marie Julie Møller; Jean-Marie Delaissé; Jacob Bastholm Olesen; Jonna Skov Madsen; Luisa Matos Canto; Troels Bechmann; Silvia Regina Rogatto; Kent Søe; Aging and menopause reprogram osteoclast precursors for aggressive bone resorption. Bone Research 2020, 8, 1-11, 10.1038/s41413-020-0102-7.
- K P McHugh; Z. Shen; T N Crotti; M R Flannery; R P O'sullivan; P E Purdue; S R Goldring; The role of cell-substrate interaction in regulating osteoclast activation: potential implications in targeting bone loss in rheumatoid arthritis. Annals of the Rheumatic Diseases 2009, 69, i83-i85, 10.1136/ard.2009.120188.
- Jianjiao Ni; Xiaofei Zhang; Juan Li; Zhiqin Zheng; Junhua Zhang; Weixin Zhao; Liang Liu; Tumour-derived exosomal lncRNA-SOX2OT promotes bone metastasis of non-small cell lung cancer by targeting the miRNA-194-5p/RAC1 signalling axis in osteoclasts. Cell Death & Disease 2021, 12, 1-10, 10.1038/s41419-021-03928-w.
- Aseel Marahleh; Hideki Kitaura; Fumitoshi Ohori; Akiko Kishikawa; Saika Ogawa; Wei-Ren Shen; Jiawei Qi; Takahiro Noguchi; Yasuhiko Nara; Itaru Mizoguchi; et al. TNF-α Directly Enhances Osteocyte RANKL Expression and Promotes Osteoclast Formation. Frontiers in Immunology 2019, 10, 2925, 10.3389/fimmu.2019.02925.
- Cheng-Ming Wei; Yi-Ji Su; Xiong Qin; Jia-Xin Ding; Qian Liu; Fang-Ming Song; Shao-Hui Zong; Jiake Xu; Bo Zhou; Jin-Min Zhao; et al. Monocrotaline Suppresses RANKL-Induced Osteoclastogenesis In Vitro and Prevents LPS-Induced Bone Loss In Vivo. Cellular Physiology and Biochemistry 2018, 48, 644-656, 10.1159/000491892.
- Steven Teitelbaum; F. Patrick Ross; Genetic regulation of osteoclast development and function. Nature Reviews Genetics 2003, 4, 638-649, 10.1038/nrg1122.
- Chengchao Song; Xiaobin Yang; Yongsheng Lei; Zhen Zhang; Wanli Smith; Jinglong Yan; Lingbo Kong; Evaluation of efficacy on RANKL induced osteoclast from RAW264.7 cells. Journal of Cellular Physiology 2018, 234, 11969-11975, 10.1002/jcp.27852.
- Sylvia Christakos; Puneet Dhawan; Angela Porta; Leila J. Mady; Tanya Seth; Vitamin D and intestinal calcium absorption. Molecular and Cellular Endocrinology 2011, 347, 25-29, 10.1016/j.mce.2011.05.038.
- Jianhong Gu; Xi-Shuai Tong; Guo-Hong Chen; Dong Wang; Yang Chen; Yan Yuan; Xue-Zhong Liu; Jian-Chun Bian; Zong-Ping Liu; Effects of 1α,25-(OH)2D3 on the formation and activity of osteoclasts in RAW264.7 cells. The Journal of Steroid Biochemistry and Molecular Biology 2015, 152, 25-33, 10.1016/j.jsbmb.2015.04.003.
- J. Wesley Pike; Sylvia Christakos; Biology and Mechanisms of Action of the Vitamin D Hormone. Endocrinology and Metabolism Clinics of North America 2017, 46, 815-843, 10.1016/j.ecl.2017.07.001.
- Tomoki Mori; Kanji Horibe; Masanori Koide; Shunsuke Uehara; Yoko Yamamoto; Shigeaki Kato; Hisataka Yasuda; Naoyuki Takahashi; Nobuyuki Udagawa; Yuko Nakamichi; et al. The Vitamin D Receptor in Osteoblast-Lineage Cells Is Essential for the Proresorptive Activity of 1α,25(OH)2D3 In Vivo. Endocrinology 2020, 161, bqaa178, 10.1210/endocr/bqaa178.
- Renata C. Pereira; Isidro B. Salusky; Richard E. Bowen; Earl G. Freymiller; Katherine Wesseling-Perry; Vitamin D sterols increase FGF23 expression by stimulating osteoblast and osteocyte maturation in CKD bone. Bone 2019, 127, 626-634, 10.1016/j.bone.2019.07.026.
- Naoyuki Takahashi; Nobuyuki Udagawa; Tatsuo Suda; Vitamin D endocrine system and osteoclasts. BoneKEy Reports 2014, 3, 495, 10.1038/bonekey.2013.229.
- Jianhong Gu; Xishuai Tong; Yang Chen; Chuang Zhang; Tianhong Ma; Saihui Li; Wenyan Min; Yan Yuan; Xuezhong Liu; Jianchun Bian; et al. Vitamin D Inhibition of TRPV5 Expression During Osteoclast Differentiation. International Journal of Endocrinology and Metabolism 2019, 17, e91583, 10.5812/ijem.91583.
- Jumpei Teramachi; Yuko Hiruma; Seiichi Ishizuka; Hisako Ishizuka; Jacques P. Brown; Laëtitia Michou; Huiling Cao; Deborah L. Galson; Mark A Subler; Hua Zhou; et al. Role of ATF7-TAF12 interactions in the vitamin D response hypersensitivity of osteoclast precursors in Paget's disease. Journal of Bone and Mineral Research 2013, 28, 1489-1500, 10.1002/jbmr.1884.
- M. Kogawa; D.M. Findlay; P.H. Anderson; G.J. Atkins; Modulation of osteoclastic migration by metabolism of 25(OH)-vitamin D3. The Journal of Steroid Biochemistry and Molecular Biology 2013, 136, 59-61, 10.1016/j.jsbmb.2012.09.008.
- Colnot, C.; Sidhu, S.S.; Poirier, F.; Balmain, N; Cellular and subcellular distribution of galectin-3 in the epiphyseal cartilage and bone of fetal and neonatal mice. Cell Mol Biol 1999, 45, 1191-1202, .
- Jane E. Aubin; F. Liu; Luc Malaval; A.K. Gupta; Osteoblast and chondroblast differentiation. Bone 1995, 17, S77-S83, 10.1016/8756-3282(95)00183-e.
- Kosei Nakajima; Dhong Hyo Kho; Takashi Yanagawa; Yosuke Harazono; Victor Hogan; Wei Chen; Rouba Ali-Fehmi; Rohit Mehra; Avraham Raz; Galectin-3 Cleavage Alters Bone Remodeling: Different Outcomes in Breast and Prostate Cancer Skeletal Metastasis.. Cancer Research 2016, 76, 1391-402, 10.1158/0008-5472.CAN-15-1793.
- Gu, J.; Kong, Q.; Wang, D.; Tong, X.; Bian, J.; Liu, X.; Yuan, Y.; Liu, Z; Deferential protein expressions and bioinformatic analysis of OC fromation regulated by vitamin D. Chinese Journal of Veterinary Science (in chinese) 2019, 39, 2207-2214, .
- Dominic Simon; Anja Derer; Fabian T. Andes; Patrick Lezuo; Aline Bozec; Georg Schett; Martin Herrmann; Ulrike Harre; Galectin-3 as a novel regulator of osteoblast-osteoclast interaction and bone homeostasis. Bone 2017, 105, 35-41, 10.1016/j.bone.2017.08.013.
- Gu, J.; Zhang, C.; Min, W.; ZHao, Y.; Li, S.; Liu, Z; Effects of 1α,25-(OH)2D3 on osteoclastogenesis and activity in mice. Chinese Journal of Veterinary Science (in chinese) 2019, 49, 593-600, .
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165-176, .
- Paola Spessotto; Francesca Maria Rossi; Massimo Degan; Raffaele Di Francia; Roberto Perris; Alfonso Colombatti; Valter Gattei; Hyaluronan–CD44 interaction hampers migration of osteoclast-like cells by down-regulating MMP-9. Journal of Cell Biology 2002, 158, 1133-1144, 10.1083/jcb.200202120.
- Saki Nakagawa; Kazuhiro Omori; Masaaki Nakayama; Hiroki Mandai; Satoshi Yamamoto; Hiroya Kobayashi; Hidefumi Sako; Kyosuke Sakaida; Hiroshi Yoshimura; Satoki Ishii; et al. The fungal metabolite (+)-terrein abrogates osteoclast differentiation via suppression of the RANKL signaling pathway through NFATc1. International Immunopharmacology 2020, 83, 106429, 10.1016/j.intimp.2020.106429.
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell 2002, 3, 889-901, .
- Masakazu Kogawa; Koji Hisatake; Gerald Atkins; David M. Findlay; Yuichiro Enoki; Tsuyoshi Sato; Peter C. Gray; Yukiko Kanesaki-Yatsuka; Paul Anderson; Seiki Wada; et al. The Paired-box Homeodomain Transcription Factor Pax6 Binds to the Upstream Region of the TRAP Gene Promoter and Suppresses Receptor Activator of NF-κB Ligand (RANKL)-induced Osteoclast Differentiation. Journal of Biological Chemistry 2013, 288, 31299-31312, 10.1074/jbc.m113.461848.
- Denis Duplat; Marlène Gallet; Sophie Berland; Arul Marie; Lionel Dubost; Marthe Rousseau; Said Kamel; Christian Milet; Michel Brazier; Evelyne Lopez; et al. The effect of molecules in mother-of-pearl on the decrease in bone resorption through the inhibition of osteoclast cathepsin K. Biomaterials 2007, 28, 4769-4778, 10.1016/j.biomaterials.2007.07.036.
- Vaishali Veldurthy; Ran Wei; Leyla Oz; Puneet Dhawan; Yong Heui Jeon; Sylvia Christakos; Vitamin D, calcium homeostasis and aging. Bone Research 2016, 4, 16041, 10.1038/boneres.2016.41.
- Riko Kitazawa; Kiyoshi Mori; Akira Yamaguchi; Takeshi Kondo; Sohei Kitazawa; Modulation of mouse RANKL gene expression by Runx2 and vitamin D3. Journal of Cellular Biochemistry 2008, 105, 1289-1297, 10.1002/jcb.21929.
- Dong Wang; Jian-Hong Gu; Yang Chen; Hong-Yan Zhao; Wei Liu; Rui-Long Song; Jian-Chun Bian; Xue-Zhong Liu; Yan Yuan; Zong-Ping Liu; et al. 1α,25-Dihydroxyvitamin D3 inhibits the differentiation and bone resorption by osteoclasts generated from Wistar rat bone marrow-derived macrophages. Experimental and Therapeutic Medicine 2015, 10, 1039-1044, 10.3892/etm.2015.2632.
- Sadaoki Sakai; Hironari Takaishi; Kenichiro Matsuzaki; Hironori Kaneko; Mitsuru Furukawa; Yoshiteru Miyauchi; Ayako Shiraishi; Keiji Saito; Akio Tanaka; Tadatsugu Taniguchi; et al. 1-Alpha, 25-dihydroxy vitamin D3 inhibits osteoclastogenesis through IFN-beta-dependent NFATc1 suppression. Journal of Bone and Mineral Metabolism 2009, 27, 643-652, 10.1007/s00774-009-0084-4.
- Junichi Kikuta; Shunsuke Kawamura; Fumie Okiji; Mai Shirazaki; Sadaoki Sakai; Hitoshi Saito; Masaru Ishii; Sphingosine-1-phosphate-mediated osteoclast precursor monocyte migration is a critical point of control in antibone-resorptive action of active vitamin D. Proceedings of the National Academy of Sciences 2013, 110, 7009-7013, 10.1073/pnas.1218799110.
- Gong, H.C.; Honjo, Y.; Nangia-Makker, P.; Hogan, V.; Mazurak, N.; Bresalier, R.S.; Raz, A; The NH2 terminus of galectin-3 governs cellular compartmentalization and functions in cancer cells. Cancer Res 1999, 59, 6239-6245, .
- Milica Velickovic; Aleksandar Arsenijevic; Aleksandar Acovic; Dragana Arsenijevic; Jelena Milovanovic; Jelena Dimitrijevic; Zeljko Todorovic; Marija Milovanovic; Tatjana Kanjevac; Nebojsa Arsenijevic; et al. Galectin-3, Possible Role in Pathogenesis of Periodontal Diseases and Potential Therapeutic Target. Frontiers in Pharmacology 2021, 12, 638258, 10.3389/fphar.2021.638258.
- Huajun Tang; Peiyue Zhang; Lianlin Zeng; Yu Zhao; Libo Xie; Bo Chen; Mesenchymal stem cells ameliorate renal fibrosis by galectin-3/Akt/GSK3β/Snail signaling pathway in adenine-induced nephropathy rat. Stem Cell Research & Therapy 2021, 12, 1-22, 10.1186/s13287-021-02429-z.
- Guodong Fu; Olena Polyakova; Ronald Chazen; Jeremy Freeman; Ian Witterick; Diagnostic Value of Galectin-3 in Distinguishing Invasive Encapsulated Carcinoma from Noninvasive Follicular Thyroid Neoplasms with Papillary-Like Nuclear Features (NIFTP). Cancers 2021, 13, 2988, 10.3390/cancers13122988.
- Weizhen Jia; Lingyu Kong; Hiroyasu Kidoya; Hisamichi Naito; Fumitaka Muramatsu; Yumiko Hayashi; Han-Yun Hsieh; Daishi Yamakawa; Daniel K. Hsu; Fu-Tong Liu; et al. Indispensable role of Galectin-3 in promoting quiescence of hematopoietic stem cells. Nature Communications 2021, 12, 1-17, 10.1038/s41467-021-22346-2.
- Carla Iacobini; Claudia Blasetti Fantauzzi; Rossella Bedini; Raffaella Pecci; Armando Bartolazzi; Bruno Amadio; Carlo Pesce; Giuseppe Pugliese; Stefano Menini; Galectin-3 is essential for proper bone cell differentiation and activity, bone remodeling and biomechanical competence in mice. Metabolism 2018, 83, 149-158, 10.1016/j.metabol.2018.02.001.
- Adriana Lepur; Michael C. Carlsson; Ruđer Novak; Jerka Dumić; Ulf J. Nilsson; Hakon Leffler; Galectin-3 endocytosis by carbohydrate independent and dependent pathways in different macrophage like cell types. Biochimica et Biophysica Acta (BBA) - General Subjects 2012, 1820, 804-818, 10.1016/j.bbagen.2012.02.018.
- Wei Liu; Chung Chi Le; Dong Wang; Di Ran; Yi Wang; Hongyan Zhao; Jianhong Gu; Hui Zou; Yan Yuan; Jianchun Bian; et al. Ca2+/CaM/CaMK signaling is involved in cadmium-induced osteoclast differentiation. Toxicology 2020, 441, 152520, 10.1016/j.tox.2020.152520.
- Xishuai Tong; Chuang Zhang; Dong Wang; Ruilong Song; Yonggang Ma; Ying Cao; Hongyan Zhao; Jianchun Bian; Jianhong Gu; Zongping Liu; et al. Suppression of AMP‐activated protein kinase reverses osteoprotegerin‐induced inhibition of osteoclast differentiation by reducing autophagy. Cell Proliferation 2019, 53, e12714, 10.1111/cpr.12714.
- Xishuai Tong; Jianhong Gu; Miaomiao Chen; Tao Wang; Hui Zou; Ruilong Song; Hongyan Zhao; Jianchun Bian; Zongping Liu; p53 positively regulates osteoprotegerin-mediated inhibition of osteoclastogenesis by downregulating TSC2-induced autophagy in vitro. Differentiation 2020, 114, 58-66, 10.1016/j.diff.2020.06.002.
- Ahmed Hasbi; Melissa L. Perreault; Maurice Y. F. Shen; Theresa Fan; Tuan Nguyen; Mohammed Alijaniaram; Tomek J. Banasikowski; Anthony A. Grace; Brian F. O'dowd; Paul Fletcher; et al. Activation of Dopamine D1-D2 Receptor Complex Attenuates Cocaine Reward and Reinstatement of Cocaine-Seeking through Inhibition of DARPP-32, ERK, and ΔFosB. Frontiers in Pharmacology 2018, 8, 924, 10.3389/fphar.2017.00924.