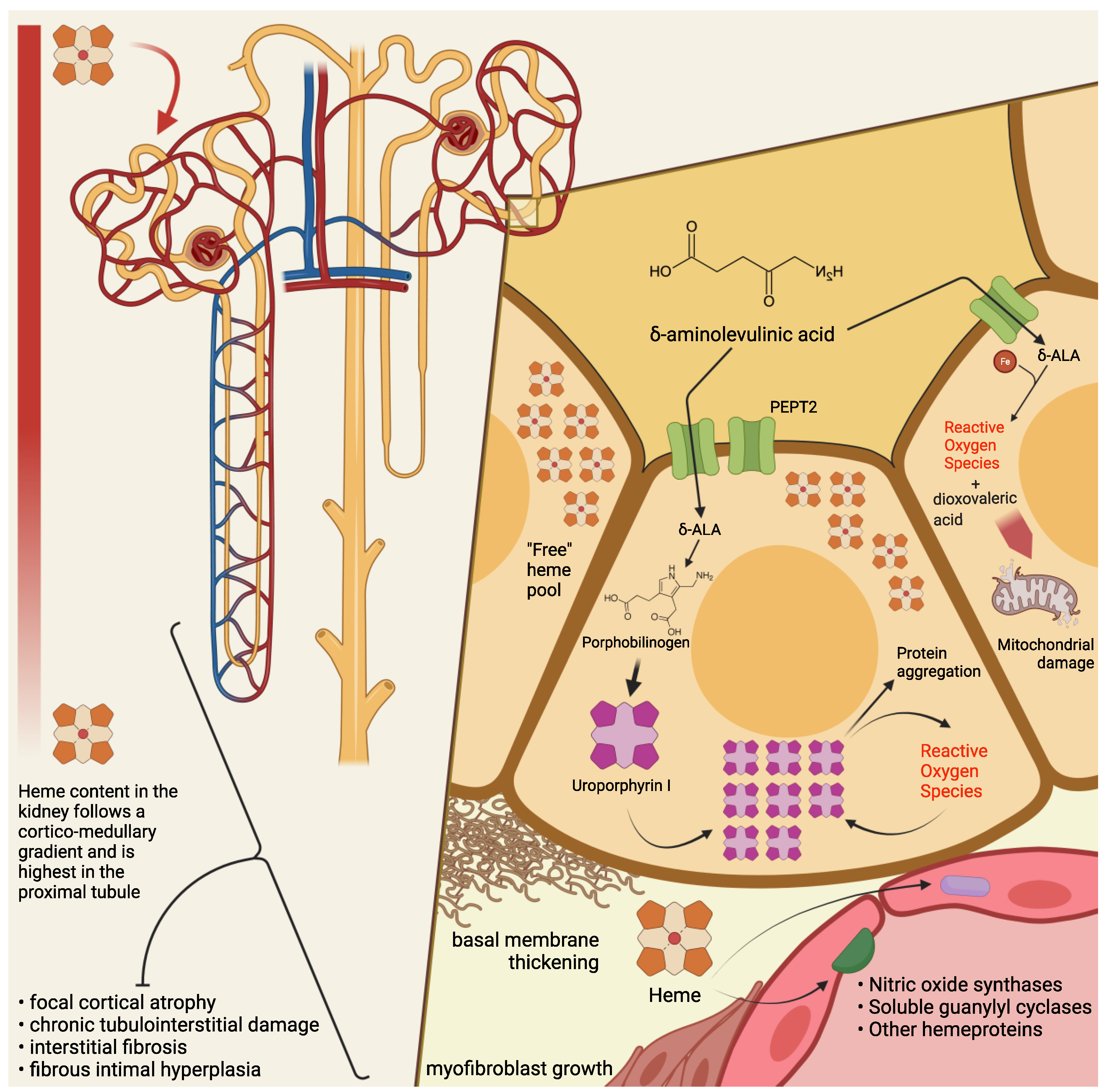
It is generally assumed that the kidney contributes to heme production as the third major synthesizing organ, after the bone marrow and the liver—which account, respectively, for 80% and 15% of total heme biosynthesis [
19]. In fact, several biochemical, ultrastructural, and fluorescence microscopy studies have suggested that the kidney is overall abundant in heme. At variance with the liver, though, the capacity for heme biosynthesis in the kidney is heterogeneously distributed and highly compartmentalized, and parallels the activity of detoxifying cytochromes and other heme-dependent functions [
20]. Thus, it has been demonstrated that heme biosynthetic activity and porphyrin concentration in the kidney follow a corticomedullary gradient: both are highest in the cortical proximal tubules, a metabolically active region particularly exposed to xenobiotics or other endogenously produced compounds [
20].
Compared to liver cells, ALAS in the kidney is somewhat more refractory to induction by porphyrinogenic stimuli. The induction
process—an initial increase in the enzyme’s activity in the cytosol and a subsequent shift into the mitochondrial matrix- seems qualitatively similar to what has been observed in the liver, but its
kinetics are much slower (i.e., hours instead of minutes) [
20]. By contrast, renal ALAS activity is promptly inhibited by heme, similarly to the liver isoform. Finally, a greater ratio of ferrochelatase-to-ALAS activity has been detected in renal compared to liver cells. Together with other pieces of evidence, these observations have led to the hypothesis that the kidney could benefit from a higher content of intracellular, regulatory “free” heme, which could also function as a protective buffer to acute heme-depleting stimuli [
20].
3. Pathogenesis of Kidney Damage in PAKD
Among several mechanisms by which ALA is thought to cause cytotoxic damage, the kidney may be particularly susceptible—at least in its most metabolically active segments—to mitochondrial ALA-induced oxidation. At the intracellular level, ALA undergoes a phosphate-catalyzed auto-enolization, and becomes an oxidizing agent; it reacts with iron and O
2 to produce superoxide anion (O
2), HO radical, and ALA radical (ALA); ALA, in the presence of oxygen, reduces iron and yields dioxo valeric acid (DOVA), a highly reactive oxidant [
36,
37]. Several pieces of evidence have been gathered concerning ALA toxicity on mitochondrial morphology, loss of transmembrane potential, and protein expression [
38,
39,
40].
Renal histopathological findings in patients with PAKD point toward chronic tubulointerstitial damage [
16,
18,
30,
41,
42,
43] and chronic fibrous intimal hyperplasia associated with focal cortical atrophy [
16]. Early autopsy reports in a South African series of patients with variegate porphyria evidenced renal tubular degeneration, more marked in distal tubules, with calcified casts [
18]. More recently, Pallet et al. [
16] described tubular atrophy, basal membrane thickening, and interstitial fibrosis; nonspecific arteriosclerotic lesions [
16] have also been observed, with arterial fibrous intimal hyperplasia in the cortex, consisting of myofibroblast growth, sclero-fibrotic tissue production and endothelial lumen narrowing. Remarkably, glomeruli seem spared from direct damage [
43], since only unspecific sclerotic and ischemic lesions have been reported [
16,
30]. Markers of ongoing fibrogenesis, such as cytoplasmic accumulation of ß-catenin and vimentin expression, [
16] have been detected in tubular sections, and mitochondrial abnormalities have been reported anecdotally [
18,
42].
Cell culture studies have shown that human endothelial cells (HUVECs), when incubated with ALA and PBG, do not appear to suffer direct damage from the porphyrin precursors [
16]. In contrast, human renal epithelial cells (HRECs) display a wide range of alterations in the presence of ALA and PBG in vitro, i.e.: activation of apoptosis, with signs of autophagy and endoplasmic reticulum stress; evidence of a proinflammatory and fibrogenic secretory milieu; morphologic and molecular changes suggestive of epithelial-to-mesenchymal transition (loss of the cuboid morphology, cell-to-cell contact, E-cadherin expression; nuclear translocation of β-catenin; increased expression of SLUG).
On electron microscopy, HRECs incubated with PBG showed accumulation of electron-dense cytosolic granules, whereas light microscopy detected yellow-brown granular aggregates, negative for Perl’s stain, and numerous cytoplasmic osmiophilic granules within the proximal tubular cells [
16]. Intriguingly, when proximal tubular cells are incubated with PBG, the latter is completely metabolized into uroporphyrinogen I and III [
16]: therefore, it has been conjectured that the observed intracellular inclusions could be aggregates of uroporphyrin obtained by the uncatalyzed polymerization and cyclisation of four PBG molecules.
It is then interesting, from a historical as well as a scientific perspective, that a few studies on acute porphyrias from the past century have reported histopathological findings suggestive of tubular deposition of porphyrins [
18,
44,
45,
46]; for instance, a case series of autopsies from patients with variegate porphyria mentioned the presence of a brown autofluorescent pigment, not staining as iron, in both casts and renal tubular cells, and detected a red-orange autofluorescence in the lumen and epithelial cells of Henle’s loop, which in the author’s experience could be possibly attributed to porphyrin deposits [
18].
As a matter of fact, consistent pieces of evidence have been gathered concerning the cell-damaging effects of light-independent porphyrin-mediated toxicity [
47]: in particular, intracellular, extralysosomal porphyrin accumulation engenders protein aggregation through noncovalent, oxygen-dependent, reversible mechanisms [
48,
49]. A particular susceptibility has been demonstrated, chiefly in hepatocytes, for intermediate filaments (nuclear laminins and cytoplasmatic keratins) [
47,
50], proteins in the endoplasmic reticulum (e.g., protein disulfide isomerase and calnexin) [
51], proteasome regulatory particles, and key glycolytic enzymes, including glyceraldehyde 3-phosphate dehydrogenase [
51]. This process could both trigger and be accelerated by the activity of other oxidizing agents (inflammation, redox reactions) [
48], so that porphyrins could precipitate the production of reactive oxygen species (ROS) and intracellular protein aggregation without prior photosensitization. Of note, uroporphyrin I is reduced by the P450 cytochrome’s family and by nicotinamide adenine dinucleotide phosphate (NADPH) in a reaction that yields a superoxide radical (O
2−) [
52,
53]. It may be tempting to speculate that similar mechanisms might take place in the cytochrome-rich renal parenchyma, contributing to the renal toxicity of high concentrations of ALA and PBG.
It must be remarked that when a mouse model of AIP was employed to investigate the effects of repeated phenobarbital-induced acute attacks on renal tissues [
54] relatively mild unspecific alterations were undisclosed, even in near-total (that is, 5/6) nephrectomized animals. No granular inclusions or signs of tubule-interstitial damage were evidenced, even though the same authors underscore the differences between the experimental setting and the patients’ condition with years of exposure to abnormal levels of porphyrin precursors [
54].
From a clinical perspective, signs of proximal tubulointerstitial insufficiency (i.e., proteinuria, impaired erythropoietin production) and of oxidative damage (increased urinary excretion of lipoperoxides), have been anecdotally signaled in porphyric patients [
41,
43]. A pattern consistent with sodium losses of tubular origin has been detected in patients with variegate porphyria [
17]. A case series reported that, during remission from acute attacks, patients with AHPs displayed signs of tubulointerstitial and hypertensive damage, such as poorly concentrated urines (hyposthenuria), and an impairment of the tubular excretory phase, as disclosed by isotopic renography. In this population, four patients had low serum erythropoietin levels, while all of them (11 with AIP, 1 with VP) had low plasma and erythrocyte vitamin B6 (pyridoxal phosphate, PLP) levels. Interestingly, all patients had significant hyperoxalaemia and hyperoxaluria, and an inverse relationship between plasma oxalic acid and erythrocyte vitamin B6 levels was found in AIP patients [
41]. Oxalic acid is a product of glyoxylic acid metabolism, whose conversion to glycine is effected by PLP-dependent transaminases [
55,
56]. Inherited excessive urinary excretion of oxalic acid (primary hyperoxaluria) is linked to an increased risk of urolithiasis (formation of calcium oxalate kidney stones) and kidney damage [
57]. Even though the efficacy of PLP supplementation in reducing oxaluria is debated [
56,
57,
58,
59,
60], AHPs patients are known to suffer from a poorer vitamin B6 status [
61,
62] compared to the general population