Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Medicine, General & Internal
Serum phosphate is crucial in the management of kidney disease, playing a major role in vascular calcification in chronic kidney failure. In the past 20 years, the role of phosphate has been profoundly reconsidered since many other molecules have been found to play important roles in phosphate homeostasis, beyond the well-known effect of parathyroid hormone (PTH) or renal function. The advent of new insights into phosphate metabolism must urge the endocrinologist to rethink the pathophysiology of widespread disorders, such as primary hyperparathyroidism.
- parathyroid hormone
- hyperparathyroidism
- phosphate
- primary hyperparathyroidism
- hypercalcemia
- hypophosphatemia
- osteomalacia
1. Introduction
Although the interpretation of serum calcium abnormalities might not be straightforward for the endocrinologist, phosphate metabolism alterations are always challenging. Understanding calcium–phosphate metabolism alterations requires deep knowledge of both bone metabolism and kidney physiology and pathology. For a long time, serum phosphate has been considered to be crucial in the management of kidney disease, playing a major role in vascular calcification in chronic kidney failure. In the past 20 years, the role of phosphate has been profoundly reconsidered since many other molecules have been found to play important roles in phosphate homeostasis, beyond the well-known effect of parathyroid hormone (PTH) or renal function. The advent of new insights into phosphate metabolism must urge the endocrinologist to rethink the pathophysiology of widespread disorders, such as primary hyperparathyroidism, or rarer endocrine disorders known to deeply affect quality of life, such as hypoparathyroidism and tumor-induced osteomalacia (TIO). Rare diseases of mineral metabolism have been and will be a precious source of new information about phosphate and minerals in the coming years.
2. Overview of Phosphate Physiology and Pathophysiology
While 99% of the total calcium of the human body is located in the bone, by comparison, approximately 85% of phosphate can be found in the skeleton. The remaining phosphate is distributed in other tissues, where this molecule is involved in several essential biological processes. Inorganic phosphate is composed essentially of two ions: dihydrogen phosphate (H2PO4−) and hydrogen phosphate (HPO42−). Only a small fraction of phosphate, approximately 10%, is complexed with cations (calcium or magnesium) or proteins [1].
Intracellular and extracellular phosphate concentrations are maintained constantly by means of sodium–phosphate cotransporters located across the plasma membrane. These cotransporters are subject to endocrine bioregulators, including PTH and FGF-23 within specific organs.
Several biological molecules are made up of organic phosphate: nucleic acids, phospholipids, carbohydrates, adenosine triphosphate (ATP), 2,3-diphosphoglycerate (2,3-DPG), and creatine phosphate, thereby making phosphate a ubiquitous player in the human body, with a special role in the energy compart. In addition, phosphate is involved in intracellular protein phosphorylation, where it serves as one of the main actors to control intracellular signal transduction and, eventually, gene regulation.
Pathophysiology of abnormal serum phosphate is dependent on alterations of intracellular or extracellular phosphorus, or both. In fact, clinical manifestations of low serum phosphate or hypophosphatemia are mainly the result of decreased intracellular phosphorus. Low 2,3-DPG causes hemoglobin to have more affinity to oxygen, preventing the release of oxygen to tissues. ATP production is impaired, thus resulting in impaired cellular function. On the other hand, high serum phosphate or hyperphosphatemia promotes calcium–phosphate deposition, known as calcification, a typically extracellular process often occurring in soft tissues or vessels. Calcium–phosphate deposition becomes pathological [2] when the calcification is able to determine the abnormal function of the tissue or specific symptoms.
Symptoms of hypophosphatemia or hyperphosphatemia might often be nonspecific and easily overlooked. If serum phosphate is low, muscle weakness and fatigue are commonly encountered. Signs of rickets or osteomalacia can also be present. More severe clinical consequences might occur if hypophosphatemia development is acute and severe, thereby leading to respiratory dysfunction, hemolysis, rhabdomyolysis, or even cardiac failure. By comparison, symptoms of chronic hyperphosphatemia are more subtle and often nonspecific, while in acute hyperphosphatemia symptoms of hypocalcemia (tingling, paresthesias, and muscle cramps) are the main clinical presentation. Chronic hyperphosphatemia might be evident with palpable, hard, subcutaneous nodules, consistent with soft tissue calcifications.
Serum phosphate is significantly higher in younger patients (e.g., infants and children) compared to adults, and abnormalities of phosphate values can be missed if the adult reference range is used to evaluate pediatric patients. Therefore, it is important to recognize hypophosphatemia when serum phosphate concentrations are below the lower limit of normal for a given age. The opposite applies to hyperphosphatemia. This biochemical feature can be explained by a positive phosphate balance in growing children; phosphate absorption in the small intestine is greater than renal phosphate excretion to allow skeletal accrual [1]. In contrast, in adults, the phosphate balance is equal to zero unless parathyroid disorders or other causes are present.
Serum phosphate must not be evaluated alone but should always be assessed under a pathophysiological perspective. Major hormones involved in phosphate physiology include 1,25(OH)2 vitamin D, parathyroid hormone (PTH), and phosphaturic peptides such as FGF-23. Figure 1 shows a simplification of normal phosphate physiology.
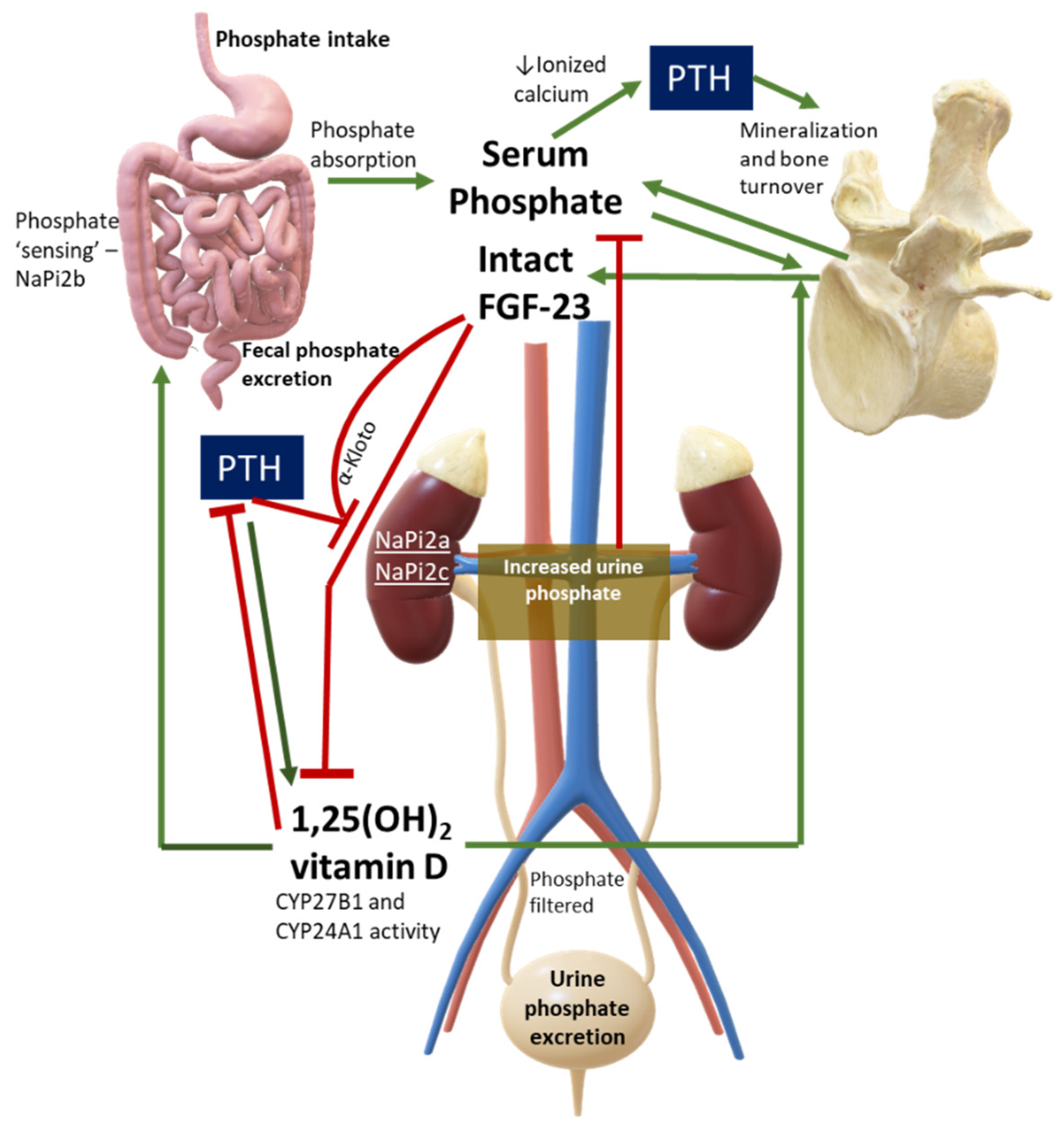
Figure 1. Phosphate Physiology. Green lines are stimulatory pathways, while red lines stand for inhibitory pathways. This figure can also be used to interpret diseases with PTH or FGF-23 excess or deficiency, or in case of abnormalities in phosphate cotransporters. Legend: CYP27B1, 1-alpha hydroxylase; CYP24A1, vitamin D 24-hydroxylase; NaPi, sodium-phosphate cotransporter.
Hypophosphatemia is a common biochemical abnormality in hospitalized patients and has been associated with poor outcomes [3]. Low serum phosphorus is seen in up to 5% of hospitalized patients [4][5]. Phosphorus is largely (60–65%) and easily absorbed in the small intestine, and its absorption is mediated by the sodium-dependent phosphate cotransporter type IIb (NaPiIIb). The higher the dietary load, the higher the intestinal absorption, with a resultant higher demand for phosphate excretion through the kidneys [6]. A recent study has assessed the biochemical response to inorganic phosphorus in healthy subjects. The study found that acute administration of phosphorus was able to increase postprandial serum phosphate levels and to stimulate the release of the parathyroid hormone, in spite of unchanged concentrations of FGF-23 in up to eight hours of observation [7]. These findings suggested that FGF-23 does not rapidly contribute to phosphate homeostasis, with PTH playing a dominant role in acute circumstances. In other studies, FGF-23 increased in response to phosphate loading, but this was observed only after several days [7].
For the clinician approaching phosphate alterations, it is important to know that approximately 80% of filtered phosphate is reabsorbed in the proximal tubule and <10% in the distal segments of the nephron [8]. Renal reabsorption of phosphate progressively increases as the filter load increases, with filtered phosphate reaching at some point a maximum tubular reabsorption rate or TmP. Because TmP depends on renal function, the correct parameter to estimate phosphate renal tubular reabsorption is TmP/GFR. If TmP/GFR is low despite low serum phosphate, this means that the kidney is wasting phosphate. This ratio should be calculated from fasting serum phosphate and creatinine and a second void urine sample. In patients with hypophosphatemia, fractional excretion of phosphate (FEP) may also be useful. FEP can be calculated from the following formula: FEP = (Urinary Phosphate × Plasma Creatinine)/(Plasma Phosphate × Urinary Creatinine). When low serum phosphorus is diagnosed, a rule of thumb is that FEP > 5% or 24-h Urinary Phosphate > 100 mg; both can be used to confirm increased renal tubular phosphate loss [2]. A diagnostic flowchart for hypophosphatemia is shown in Figure 2 to guide classification of the underlying disorder. Of note, intestinal malabsorption of phosphorus, phosphorus redistribution into cells, and increased loss through the kidney are the three most common mechanisms behind hypophosphatemia.
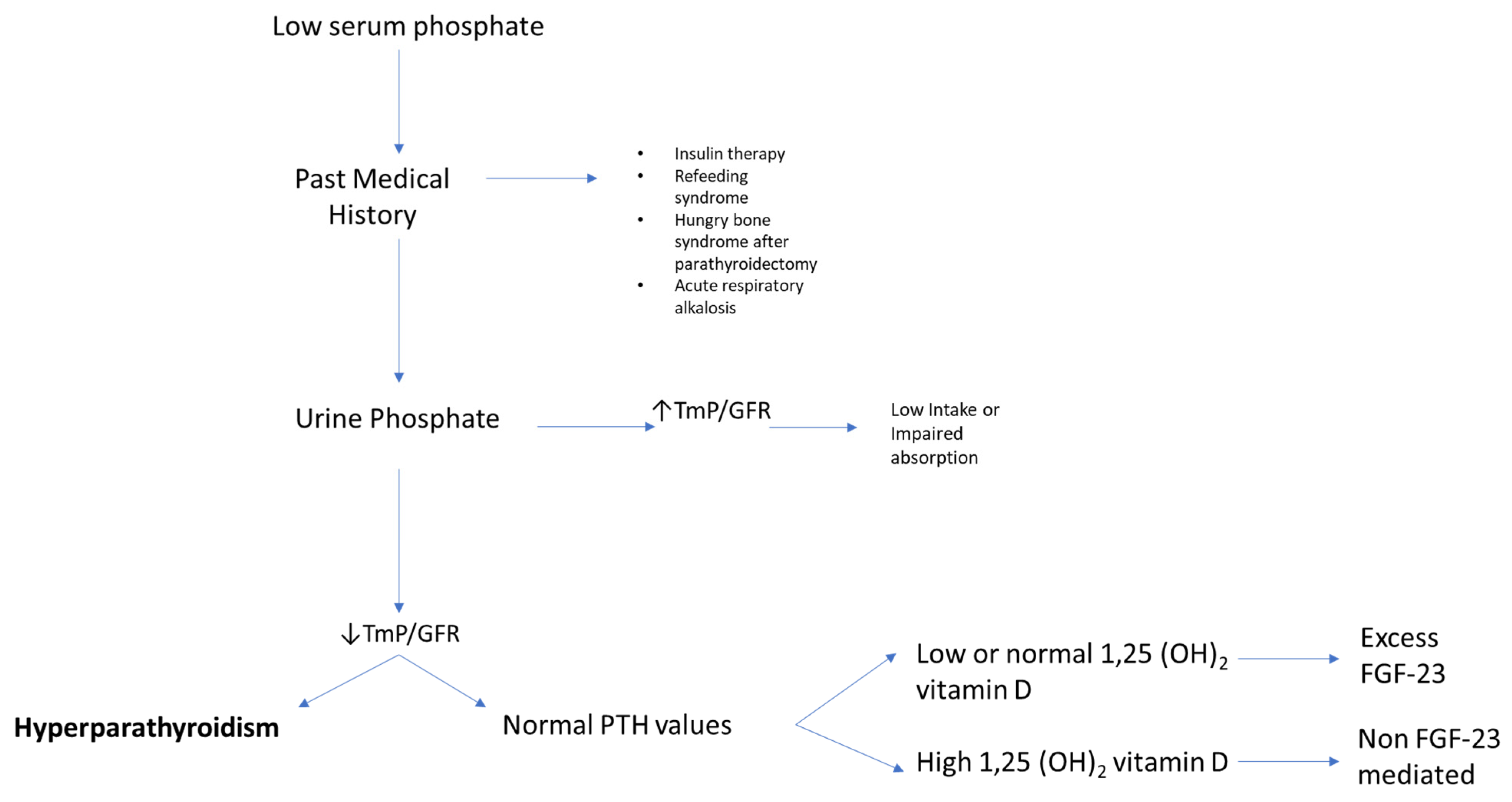
Figure 2. Flowchart for evaluating patients presenting with low serum phosphate (hypophosphatemia). High TmP/GFR is indicative of high renal reabsorption of phosphate, which is appropriate in case of low intake or malabsorption. When TmP/GFR is inappropriately low in the setting of hypophosphatemia, this suggests renal phosphate wasting. Legend: TmP/GFR, maximum tubular reabsorption of phosphate corrected for glomerular filtration rate.
3. Why Measure Serum Phosphate?
Low mineralization of the skeleton or ectopic calcification are opposite examples of how derangements of phosphate physiology might easily affect human tissue integrity. Serum phosphate abnormalities almost always hide an underlying health issue. The next few paragraphs will aim to present the importance of phosphate levels in parathyroid disorders, both in primary hyperparathyroidism and in hypoparathyroidism. Tumor-induced osteomalacia will then be discussed with a focus on phosphate and PTH interactions.
4. Phosphate in Primary Hyperparathyroidism
An early study from Columbia University [9] investigated the interactions between oral phosphate and bone health indices, including parathyroid hormone levels. Short term oral phosphate administration was shown to decrease serum calcium levels and raise PTH levels within hours, and to subsequently cause a stable rise of PTH from the third day. The presumed mechanism was attributed to decreased ionized calcium, leading to increased PTH secretion. The results of this study, conducted in 1986, are still current. Calcium and phosphate relationships have been recently reappraised in patients with primary hyperparathyroidism, with more studies focusing on ratios between minerals rather than the single mineral alone. Nowadays, it is essential to recognize early or milder phenotypes of primary hyperparathyroidism, e.g., normocalcemic primary hyperparathyroidism (NHPT), where, by definition, only the parathyroid hormone is altered, and the measurement of serum calcium alone probably becomes less useful because it is persistently within normal limits. Adding serum phosphate to the diagnostic flowchart of primary hyperparathyroidism could confirm or exclude an NHPT diagnosis, at least according to some studies [10][11].
In traditional primary hyperparathyroidism, serum phosphate is usually in the lower range of normal. In about one-quarter of patients, it is frankly below normal. It is unusual to find patients with primary hyperparathyroidism and serum phosphate values above 3.5 mg/dL in the absence of significant renal insufficiency [12]. The calcium/phosphate ratio might be used as a new index to suspect or confirm a diagnosis of PHPT [10][11]. Madeo and colleagues proposed the specific cut-point for the serum Ca/P ratio of 3.5 when Ca and P were measured in mg/dL [10]. This index had a sensitivity of 89% and specificity of 91% for detecting patients with both classical and normocalcemic primary hyperparathyroidism. In normocalcemic patients, the sensitivity of the Ca/P ratio was lower at 67%, although with comparable specificity. The authors concluded that the Ca/P ratio could be used in primary care settings or during high-volume screenings of patients because it could avoid the unnecessary measurement of PTH in many patients. However, a significant proportion of patients with NHPT might easily be missed if this ratio were used as a screening tool. This is probably due to a milder phenotype of normocalcemic hyperparathyroidism, where serum phosphate values are usually greater compared with traditional PHPT [13]. It is noteworthy that the Ca/P ratio had very high negative predictive values of 88–95%, suggesting that it could be a more reliable tool to exclude rather than confirm milder phenotypes of PHPT.
Another index, the PFindex, proposed by Guo and colleagues [14], is instead calculated from (serum calcium × PTH)/serum phosphate, with calcium and phosphate reported in mmol/L, and PTH in pg/mL. These authors retrospectively evaluated 128 patients, either with PHPT or normocalcemic PHPT, undergoing parathyroid surgery. A PFindex >34 was able to discriminate primary hyperparathyroidism from vitamin D-deficient secondary hyperparathyroidism with a sensitivity of 96.9% and a specificity of 97.6%.
Other tools such as Chloride to Phosphate Ratio (Cl:PO4 ratio) seem less useful as diagnostic laboratory tests in mild PHPT. A recent study evaluated 226 patients undergoing parathyroidectomy. Of these, 166 patients had serum calcium less than 10.4 mg/dL and were hyperchloremic (serum chloride > 106 mmol/L). Although intriguing, the sensitivity and specificity of the Cl:PO4 ratio were 58.4% and 28.6%, respectively [15].
Studies on the clinical role of FGF-23 in PHPT are scarce, although this hormone might have a potential role in the severity of clinical manifestations or symptoms in these patients. Twenty-nine PHPT patients were evaluated prospectively before and after parathyroidectomy [16]. The authors found that 1.25(OH)2 vitamin D levels correlated with FGF-23 levels both preoperatively and postoperatively, although they could not find any other significant relationship with calcium, phosphate, and PTH. Another recent study has assessed FGF-23 in PHPT patients [17]. Seventeen hypercalcemic PHPT patients were evaluated before parathyroidectomy and 6 months after surgery. Before surgery FGF-23 levels were higher compared with nine age-matched controls. FGF-23 levels decreased significantly after parathyroidectomy but remained higher compared with controls. These data suggest that FGF-23 is abnormal in PHPT, but its clinical significance and implications deserve further study.
If the interpretation of abnormalities in phosphate metabolism in normal circumstances is challenging, it could be even more challenging in patients with PHPT, which is the third most common endocrine disorder [18]. In fact, underlying phosphate disturbances may be missed in PHPT. For example, a study from the Mayo Clinic [19] aimed to characterize a group of 50 patients with coexisting PHPT and sarcoidosis, which can both present with hypercalcemia. The study found that mean serum phosphate levels were 3.3 ± 0.6 mg/dL in this cohort, and that patients with active sarcoidosis had higher serum Angiotensin Converting Enzyme (ACE) levels, lower PTH levels (60 ± 24 vs. 96 ± 41 pg/mL), and lower phosphate levels (2.7 ± 0.6 vs. 3.2 ± 0.5 mg/dL), as compared with patients with inactive sarcoidosis. Of note, serum phosphate levels were unexpectedly lower in patients with active sarcoidosis. The authors concluded that there could be other mechanisms causing lower serum phosphate in this population.
A very recent study rediscovered a new pathogenetic role for phosphate in primary hyperparathyroidism [20]. Castellano et al. evaluated a series of 472 consecutive patients with PHPT and aimed to establish a relationship between phosphate levels and clinical manifestations. These authors found that 41.9% of the patients presented with low serum phosphate mildly decreased (between 2 and 2.5 mg/dL) in the vast majority of cases (84.9%). Patients with a more pronounced phenotype of PHPT had a higher prevalence of hypophosphatemia and nephrolithiasis, but not osteoporosis. The authors suggested that moderate hypophosphatemia (1–1.9 mg/dL) in PHPT should be considered as a supportive, inexpensive, and easily available means to identify asymptomatic PHPT patients who may benefit from surgery. Cinacalcet, compared to placebo, can also increase serum phosphate levels in patients with PHPT [21], but its impact on mineralization and bone density is probably neutral in these patients [22].
This entry is adapted from the peer-reviewed paper 10.3390/ijms222312975
References
- Christov, M.; Jüppner, H. Phosphate homeostasis disorders. Best Pract. Res. Clin. Endocrinol. Metab. 2018, 32, 685–706.
- Clarke, B.L. Phosphorus Disorders: Hypophosphatemic Rickets. In Book Metabolic Bone Diseases, A Case-Baesd Approach, 1st ed.; Camacho, P.M., Ed.; Springer Nature Switzerland: Cham, Switzerland, 2019; Volume 3, pp. 83–98.
- Brunelli, S.M.; Goldfarb, S. Hypophosphatemia: Clinical consequences and management. J. Am. Soc. Nephrol. 2007, 18, 1999–2003.
- Halevy, J.; Bulvik, S. Severe hypophosphatemia in hospitalized patients. Arch. Intern. Med. 1988, 148, 153–155.
- Larsson, L.; Rebel, K.; Sörbo, B. Severe hypophosphatemia—A hospital survey. Acta Med. Scand. 1983, 214, 221–223.
- Imel, E.A.; Econs, M.J. Approach to the hypophosphatemic patient. J. Clin. Endocrinol. Metab. 2012, 97, 696–706.
- Volk, C.; Schmidt, B.; Brandsch, C.; Kurze, T.; Schlegelmilch, U.; Grosse, I.; Ulrich, C.; Girndt, M.; Stangl, G.I. Acute effects of an inorganic phosphorus additive on mineral metabolism and cardiometabolic risk factors in healthy subjects . J. Clin. Endocrinol. Metab. 2021.
- Tenenhouse, H.S. Cellular and molecular mechanisms of renal phosphate transport. J. Bone Miner. Res. 1997, 12, 159–164.
- Silverberg, S.J.; Shane, E.; Clemens, T.L.; Dempster, D.W.; Segre, G.V.; Lindsay, R.; Bilezikian, J.P. The effect of oral phosphate administration on major indices of skeletal metabolism in normal subjects. J. Bone Miner. Res. 1986, 1, 383–388.
- Madeo, B.; Kara, E.; Cioni, K.; Vezzani, S.; Trenti, T.; Santi, D.; Simoni, M.; Rochira, V. Serum Calcium to Phosphorous (Ca/P) Ratio Is a Simple, Inexpensive, and Accurate Tool in the Diagnosis of Primary Hyperparathyroidism. JBMR Plus 2018, 2, 109–117.
- Madeo, B.; De Vincentis, S.; Repaci, A.; Altieri, P.; Vicennati, V.; Kara, E.; Vescini, F.; Amadori, P.; Balestrieri, A.; Pagotto, U.; et al. The calcium-to-phosphorous (Ca/P) ratio in the diagnosis of primary hyperparathyroidism and hypoparathyroidism: A multicentric study. Endocrine 2020, 68, 679–687.
- Silverberg, S.J.; Bilezikian, J.P. Asymptomatic Primary Hyperparathyroidism. In The Parathyroids, Basic and Clinical Concepts, 3rd ed.; Bilezikian, J.P., Ed.; Elsevier: Amsterdam, The Netherlands, 2015; Section II; pp. 317–330.
- Palermo, A.; Naciu, A.M.; Tabacco, G.; Falcone, S.; Santonati, A.; Maggi, D.; D’Onofrio, L.; Briganti, S.I.; Castellitto, D.; Casini, A.; et al. Clinical, Biochemical, and Radiological Profile of Normocalcemic Primary Hyperparathyroidism. J. Clin. Endocrinol. Metab. 2020, 105, dgaa174.
- Guo, Y.; Wang, Q.; Lu, C.; Fan, P.; Li, J.; Luo, X.; Chen, D. New parathyroid function index for the differentiation of primary and secondary hyperparathyroidism: A case-control study. BMC Endocr. Disord. 2020, 20, 5.
- Wright, C.; King, D.; Small, M.; Gibson, C.; Gardner, R.; Stack, B.C., Jr. The Utility of the Cl:PO4 Ratio in Patients with Variant Versions of Primary Hyperparathyroidism. Otolaryngol. Head Neck Surg. 2021, 164, 308–314.
- Hassani, S.; Afkhamizadeh, M.; Teimouri, A.; Najaf Najafi, M.; Vazifeh Mostaan, L.; Mohebbi, M. Evaluation of Serum Level of FGF23 and 1,25(OH)2D3 in Primary Hyperparathyroidism Patients Before and After Parathyroidectomy. Int. J. Gen. Med. 2020, 13, 289–295.
- Sykała, M.; Szumowski, P.; Mojsak, M.; Abdelrazek, S.; Żukowski, Ł.; Lipińska, D.; Juchnicka, I.; Kozłowska, G.; Szelachowska, M.; Krętowski, A.; et al. Assessment of Clinical Utility of Assaying FGF-23, Klotho Protein, Osteocalcin, NTX, and Sclerostin in Patients with Primary Hyperparathyroidism. J. Clin. Med. 2021, 10, 3089.
- Wermers, R.A.; Khosla, S.; Atkinson, E.J.; Achenbach, S.J.; Oberg, A.L.; Grant, C.S.; Melton, L.J., III. Incidence of primary hyperparathyroidism in Rochester, Minnesota, 1993–2001: An update on the changing epidemiology of the disease. J. Bone Miner. Res. 2006, 21, 171–177.
- Lim, V.; Clarke, B.L. Coexisting primary hyperparathyroidism and sarcoidosis cause increased angiotensin-converting enzyme and decreased parathyroid hormone and phosphate levels. J. Clin. Endocrinol. Metab. 2013, 98, 1939–1945.
- Castellano, E.; Attanasio, R.; Boriano, A.; Pellegrino, M.; Borretta, G. Serum Phosphate: A Neglected Test In The Clinical Management Of Primary Hyperparathyroidism . J. Clin. Endocrinol. Metab. 2021, dgab676.
- Khan, A.; Bilezikian, J.; Bone, H.; Gurevich, A.; Lakatos, P.; Misiorowski, W.; Rozhinskaya, L.; Trotman, M.L.; Tóth, M. Cinacalcet normalizes serum calcium in a double-blind randomized, placebo-controlled study in patients with primary hyperparathyroidism with contraindications to surgery. Eur. J. Endocrinol. 2015, 172, 527–535.
- Vestergaard, P. Current pharmacological options for the management of primary hyperparathyroidism. Drugs 2006, 66, 2189–2211.
This entry is offline, you can click here to edit this entry!