Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Agriculture, Dairy & Animal Science
|
Allergy
Sleep apnea syndrome (SAS) is a prevalent disorder characterized by recurrent apnea or hypoxia episodes leading to intermittent hypoxia (IH) and arousals during sleep.
- intermittent hypoxia
- sleep apnea syndrome
- Reg family
- diabetes
- hepatokine
- adipokine
1. Intermittent Hypoxia and Hepatokines
It has long been known that there is a close relationship between glucose intolerance and liver disease. In the insulin-resistant state, lipolysis in adipose tissue is enhanced, leading to increased FFA in the blood and increased FFA influx into the liver. The pancreatic β cells compensatively increase insulin synthesis and secretion, resulting in hyperinsulinemia and increased synthesis of new fatty acids in the liver, leading to a nonalcoholic fatty liver disease (NAFLD). The excessive accumulation of triglycerides in the liver induces oxidative stress and inflammation, leading to the development of nonalcoholic steatohepatitis (NASH). In addition, abnormalities in hepatic lipid metabolism caused by insulin resistance lead to increased secretion of atherosclerosis-inducing lipoprotein abnormalities, inflammatory cytokines, and blood clot promoting factor, which promote atherosclerosis and create high-risk conditions for cardiovascular diseases [1][2].
Obstructive sleep apnea syndrome (OSAS) has recently been linked to NAFLD, the most common chronic liver disease in the world, which is found in about 25% of the general adult population and up to 75% of obese people [3][4][5]. According to some studies, chronic IH in patients with OSAS may itself cause liver injury, inflammation, and fibrosis, promoting the development of NAFLD and progression from lipidosis to steatohepatitis, cirrhosis, and hepatocellular carcinoma. In patients with NAFLD, IH may cause liver disease indirectly by promoting inflammation and insulin resistance and directly by promoting the production of inflammatory cytokines and metabolic abnormalities in hepatocytes [3][6][7][8][9][10][11].
A few studies investigated the relationship between IH and cytokines released from the liver (Figure 1). Briancon-Marjollet et al. investigated the respective effects of obesity and IH on inflammatory and cardiometabolic state in rats by exposing lean and obese rats to normoxic conditions or chronic IH and evaluating their serum leptin (LEP), adiponectin (ADIPOQ), hepatic cytokines, nuclear factor-κB (NF-κB) activity, and cardiac endothelin-1 levels. The results showed that the levels of IL-6 and TNF-α in the liver were elevated in lean rats exposed to IH [12]. Mesarwi et al. published the following research findings on the association of IH with the progression of NAFLD. NAFLD with steatohepatitis was induced in mice with the hepatocyte-specific deletion of hypoxia-inducible factor (Hif)1α and in wild-type control mice. The WD and IH groups resembled the histological characteristics of pediatric NASH. In addition, the levels of IL-1β, IL-6, and IL-18 were elevated in the WD and IH groups, suggesting that they promote the inflammatory response in the liver [10].
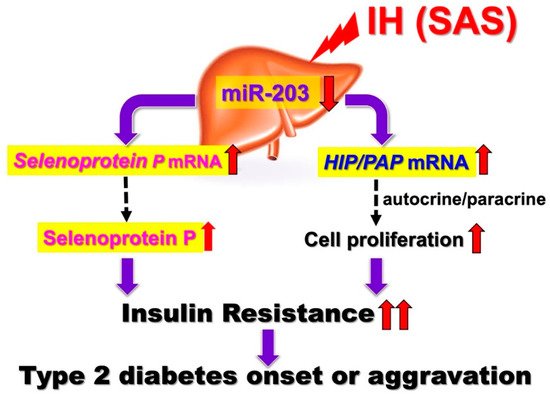
Figure 1. IH-induced upregulation of SeP and HIP/PAP via downregulation of miR-203. IH stress upregulates the levels of SeP in human hepatocytes to accelerate insulin resistance and upregulates the levels of HIP/PAP mRNAs to cause such hepatocytes to proliferate, via the miR-203 mediated mechanism. [13]. In SAS patients, it is suggested that the upregulation of SeP plays a role in worsening insulin resistance. In addition, overexpression of HIP/PAP could cause such hepatocytes to proliferate in SAS patients, leading to decreased insulin sensitivity. Red up and down arrows mean increment and decrease, respectively.
The regenerating gene (Reg) was identified in regenerating islets [14][15][16] and a Reg gene product—Reg protein—acts as a growth factor and promotes cell proliferation and regeneration [14][17][18]. In humans, five functional Reg family genes (REG Iα, REG Iβ, REG III, HIP/PAP, and REG IV) have been isolated. For several cells, it has been suggested that Reg family proteins are involved in cellular proliferation [14][16]. Ota et al. reported that IH stress stimulates pancreatic β cell proliferation via the up-regulation of Reg family mRNAs and may cause hyperinsulinemia, which makes patients more obese [19]. However, the direct effects of IH on hepatocyte proliferation and the IH-induced changes in Reg family gene expression in hepatocytes remain unknown. HepG2 cell proliferation was significantly increased by IH. Furthermore, the RNA interference of HIP/PAP inhibited HepG2 cell proliferation measured by WST-8 assay, whereas the interference of other REG family genes such as REG Iα did not. These results indicate that HIP/PAP, upregulated by IH, works as an autocrine/paracrine growth factor in hepatocyte proliferation. In addition, the IH-induced upregulation of SeP and HIP/PAP was revealed to be mediated via the downregulation of miR-203 in hepatocytes [13]. In SAS patients, it is suggested that the upregulation of SeP plays a role in worsening insulin resistance (Figure 1).
In addition, the overexpression of HIP/PAP could cause such hepatocytes to proliferate in SAS patients, leading to decreased insulin sensitivity. Moreover, Takeda et al. recently reported that the expression of renin in juxtaglomerular cells was significantly increased in response to IH stimulation via the downregulation of miR-203 [20]. The most common complications in SAS patients are hypertension and diabetes, and IH, caused by SAS, reduced miR-203 in hepatocytes [13] and juxtaglomerular cells [20], resulting in increased Sep in hepatocytes, a diabetogenic hepatokine, and renin in juxtaglomerular cells, which induces hypertension, simultaneously.
2. Intermittent Hypoxia and Adipokines
Adipose tissue is a complex tissue composed of preadipocytes, adipocytes, and interstitial vascular cells and is one of the major organs that contribute to worsening insulin resistance through inflammation and subsequent impaired function. It has long been known that the inflammation and dysfunction of adipose tissue play a pivotal role in the pathogenesis of diabetes. Adipocytes not only convert excess energy into triglycerides and store them but also express and secrete adipokines as endocrine organs to control and regulate the metabolism of the whole body. Numerous studies have been undertaken on the mechanisms of adipose tissue dysfunction in obese patients. In obesity, adipocytes hyperplasia and hypertrophy may occur [21][22][23]; however, obesity not only leads to increased fat storage but also to the abnormal function of adipose tissue as an endocrine organ. Adipocyte hypertrophy leads to functional abnormalities and inflammation, resulting in insulin resistance and immune cell infiltration, localized hypoxia, and fibrosis. Following the infiltration of CD8-positive cytotoxic T cells and the phenotypic change to M1 macrophages, inflammatory adipokines including TNF-α, IL-6, and RETN are produced, leading to insulin resistance [21][22][24][25]. Additionally, hypoxia is thought to be an important factor in adipose tissue dysfunction in obesity. Continued adipocyte hypertrophy leads to local hypoxia and the activation of the hypoxia-inducible transcription factors, in particular HIF-1, which is regarded as the key molecule for signaling the cellular response to low oxygen levels, thus contributing to dysfunctions and metabolic disorders in adipocytes [26].
The mechanism of inflammatory cell infiltration and other mechanisms (such as sympathetic activation and the exacerbation of oxidative stress) are assumed to be common pathways. Due to sympathetic activation, IH cause an increase in blood catecholamines [27], which in turn causes lipolysis and produces FFA, leading to insulin resistance in skeletal muscle and liver cells [28][29]. The chronic elevation of FFA levels has an adverse effect on glucose homeostasis, called lipotoxicity [30]. In addition, in the IH environment, HIF activation by adipose tissue hypoxia has been reported to worsen insulin sensitivity [31], and HIF-1α protein expression in the blood is reportedly significantly elevated in SAS patients [32][33]. Some reports suggest that HIF-1 is activated in sustained hypoxia (SH), but not in IH, and that inflammatory pathways are selectively activated [34]. In the IH environment, both HIF-1 and NF-κB are activated, leading to changes in gene expression. Since HIF-1 is a direct target, such as being transcriptionally regulated by NF-κB, and TNF-α is also regulated by NF-κB, it is possible that crosstalk between HIF-1 and NF-κB is associated with cytokine abnormalities under IH, but the detailed mechanism is not known [34][35]. As described above, there are several possible mechanisms by which IH induces insulin resistance and not all of them have been elucidated. We conducted research focusing on adipokines.
Adipokines are multiple hormones, cytokines, chemokines, and other proteins secreted by adipocytes in white adipose tissue [22]. Some adipokines are specific to adipocytes, and some are not. More than 50 types of adipokines have been identified to date, including TNF-α and IL-6, which are known to be factors that increase the risk of diabetes and cardiovascular disease [36][37][22]. The most common adipokines involved in insulin resistance include leptin (LEP), which is involved in appetite control and hypersensitivity to insulin, ADIPOQ, which promotes glucose uptake in skeletal muscle and liver, resistin (RETN), which exacerbates insulin resistance, and C-C motif chemokine ligand 2 (CCL2), which is involved in immunoregulatory and inflammatory processes and is a critical factor for monocyte infiltration [36][22][26][38][39], ANGPTL, which regulates glucose homeostasis, lipid metabolism, and insulin sensitivity [40][41], plasminogen activator inhibitor-1 (PAI-1), and vascular endothelial growth factor (VEGF), which is linked to the inflammatory response [26].
The detailed mechanism of association adipokines with SAS remains unclear. Recently, it was reported that FFA and inflammatory mediators such as TNF-α in serum are elevated in SAS patients and mice in an experimental IH environment, and several in vivo studies reported that IH causes adipose tissue inflammation and insulin resistance [31], suggesting that IH may enhance inflammation and dysfunction in adipose tissue [42][21].
As shown in Figure 2, the abnormal secretion of TNF-α, IL-6, and CCL2, etc., is suggested in adipose tissue under IH conditions, including in our study. Ge et al. have shown that macrophages accumulate in adipose tissue when lean mice are exposed to IH and in obese mice [43]. CCL2 may induce monocytes into adipose tissue and induce inflammation in an IH environment. Although not all SAS patients are obese, exposure to IH may exacerbate insulin resistance by a mechanism similar to that found with obesity under conditions involving adipokines. Recently, Akasaka et al. reported that advanced glycation endproduct, high-mobility group box 1, and lipopolysaccharide in pregnant women upregulate the expression of IL-6 and CCL2 in adipocytes, leading to systemic inflammation such as preeclampsia/hypertensive disorders of pregnancy [44]. As an association between sleep-disordered breathing, gestational hypertension, and preeclampsia has been demonstrated [45][46][47], CCL2 expressed and secreted from patient adipocytes may link between SAS and hypertensive disorders of pregnancy/gestational diabetes.
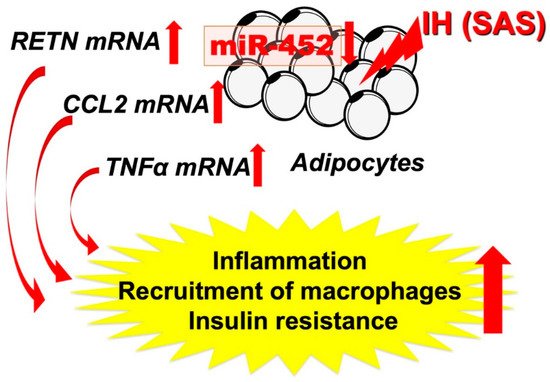
Figure 2. IH-induced increases in RETN, TNFα, and CCL2 via downregulates the miR-452 in adipocytes. It is suggested that, in SAS patients, the upregulation of RETN, TNFα, and CCL2 [48] may induce a proinflammatory phenotype of the adipose tissue, leading to the development of insulin resistance and reduced insulin sensitivity, and miR-452 could play crucial roles in the regulation of these gene expressions. Red up and down arrows mean increment and decrease, respectively.
3. Intermittent Hypoxia and Myokines
Myokines, a comprehensive term for cytokines secreted by skeletal muscle, are released from skeletal muscle during muscle contraction [1][49]. The roles of myokines are diverse and include metabolic regulation, anti-inflammatory effects, and the regulation of skeletal muscle mass during injury regeneration [1][50]. Myokines include proteins, microRNA (miRNA)s, and exosomes, and hundreds of them have been found, but only a few of their biological functions have been elucidated. These myokines exhibit autocrine/paracrine and endocrine effects and may mediate the beneficial effects of exercise on other major organs involved in the regulation of energy homeostasis [50][51]. Adipose tissue appears to be an important target for myokines, which regulate energy flux and fuel supply during muscle contraction [1][50]. IL-6 was the first myokine to be discovered, and the level of circulating IL-6 secreted by skeletal muscle is markedly elevated during exercise and muscle contraction. Some clinical studies have shown that IL-6 is needed to reduce visceral adipose tissue during exercise [52], which in turn may lead to improved insulin resistance. Park T.J. et al. found that myonectin (MN) inhibits adipogenesis in 3T3-L1 preadipocytes by downregulating the expression of adipogenic transcription factors such as CCAAT/enhancer binding protein (C/EBP) α, β, and peroxisome proliferator-activated receptor (PPAR) γ. Furthermore, they showed that MN has an inhibitory effect on adipogenesis through the regulation of the p38 mitogen-activated protein kinase (MAPK) pathway and C/EBP homologous protein (CHOP). These results suggest that MN may be a new therapeutic target for obesity prevention [53] and required for metabolic homeostasis [54]. In addition, there are various other myokines that are secreted by skeletal muscle and influence pancreatic, hepatic, and adipose tissue and that are thought to affect glucose tolerance through various mechanisms. Many myokines act not only on other organs but also on themselves (autocrine), leading to the hypertrophy of skeletal muscle and increased insulin sensitivity (increased glucose uptake). Myokines such as myostatin, on the other hand, act on the self (autocrine), causing a decrease in skeletal muscle mass, worsening insulin resistance and promoting fat deposition in the liver [1]. Myokines are involved in the anti-inflammatory effects of physical activity and counteract the metabolic abnormalities of insulin resistance and diabetes [55][56]. Therefore, abnormalities in myokine secretion and function may have a direct influence on the worsening of insulin resistance. However, the mechanism by which myokines affect insulin resistance has not been fully elucidated [50][55][56][57].
Moreover, few studies have examined the direct effect of IH on myokine secretion. Otaka et al. examined the role of MN in myocardial ischemic injury by using knockout mice subjected to I/R conditions and found that MN increased cardiac dysfunction, apoptosis, and inflammatory gene expression more than with wild-type mice. On the other hand, transgenic mice overexpressing MN showed less myocardial damage after I/R, indicating that MN functions as an endurance exercise-induced myokine and ameliorates acute myocardial ischemic injury by suppressing apoptosis and inflammation in the heart [58]. I/R is characterized by a limited blood supply to the organs and subsequent tissue damage as a result of recovery. Many studies have shown that inflammation associated with I/R injury can exacerbate myocardial damage [59], which may cause a condition experimentally similar to IH. On the basis of these studies [60], we suspected that IH had a direct effect on myokine secretion and conducted the following experiments. IH exposure increases IL-8, ON (osteonectin), and MN mRNA levels in mammalian muscle cells and octamer binding transcription factor 1 (OCT1) is a key factor for the IH-induced upregulation of IL-8 and MN mRNA expression levels and that nuclear factor erythroid 2-related factor 2 (NRF2) serves as an essential factor for the IH-induced upregulation of ON mRNA expression [61] (Figure 3). No other study has examined the direct relationship between myokine and IH; therefore, further research is desirable.
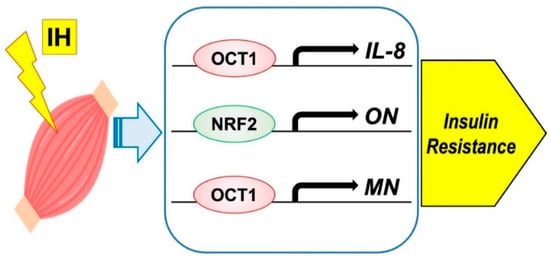
Figure 3. Possible mechanism on IH-induced insulin resistance via myokine expression in muscle cells. IH significantly increased the mRNA levels of IL-8, ON, and MN. The promoters contain consensus transcription factor binding sequences Figure 1 in IL-8 and MN, and for NRF2 in ON, respectively.
Cardiomyokines are proteins secreted by a healthy or a diseased heart, and they perform a beneficial autocrine/paracrine function [62][63][64]. IH significantly increased the mRNA levels of Reg IV and hepatocyte growth factor (Hgf) in the cardiomyocytes, and the gene expression of Reg IV and Hgf was increased via downregulation of the miR-499 level in IH-treated cardiomyocytes. It is suggested that, in SAS patients, the upregulation of Reg IV and Hgf may function against the apoptosis of cardiomyocytes, leading to the maintenance of cardiac functions, and that miR-499 could play crucial roles in the regulation of these gene expressions [65].
This entry is adapted from the peer-reviewed paper 10.3390/ijms222312898
References
- de Oliveira Dos Santos, A.R.; de Oliveira Zanuso, B.; Miola, V.F.B.; Barbalho, S.M.; Santos Bueno, P.C.; Flato, U.A.P.; Detregiachi, C.R.P.; Buchaim, D.V.; Buchaim, R.L.; Tofano, R.J.; et al. Adipokines, myokines, and hepatokines: Crosstalk and metabolic repercussions. Int. J. Mol. Sci. 2021, 22, 2639.
- Eslam, M.; Valenti, L.; Romeo, S. Genetics and epigenetics of NAFLD and NASH: Clinical impact. J. Hepatol. 2018, 68, 268–279.
- Mesarwi, O.A.; Loomba, R.; Malhotra, A. Obstructive sleep apnea, hypoxia, and nonalcoholic fatty liver disease. Am. J. Respir. Crit. Care Med. 2019, 199, 830–841.
- Aron-Wisnewsky, J.; Clement, K.; Pepin, J.L. Nonalcoholic fatty liver disease and obstructive sleep apnea. Metabolism 2016, 65, 1124–1135.
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922.
- Nobili, V.; Cutrera, R.; Liccardo, D.; Pavone, M.; Devito, R.; Giorgio, V.; Verrillo, E.; Baviera, G.; Musso, G. Obstructive sleep apnea syndrome affects liver histology and inflammatory cell activation in pediatric nonalcoholic fatty liver disease, regardless of obesity/insulin resistance. Am. J. Respir. Crit. Care Med. 2014, 189, 66–76.
- Kang, H.H.; Kim, I.K.; Lee, H.I.; Joo, H.; Lim, J.U.; Lee, J.; Lee, S.H.; Moon, H.S. Chronic intermittent hypoxia induces liver fibrosis in mice with diet-induced obesity via TLR4/MyD88/MAPK/NF-kB signaling pathways. Biochem. Biophys. Res. Commun. 2017, 490, 349–355.
- Jullian-Desayes, I.; Trzepizur, W.; Boursier, J.; Joyeux-Faure, M.; Bailly, S.; Benmerad, M.; Le Vaillant, M.; Jaffre, S.; Pigeanne, T.; Bizieux-Thaminy, A.; et al. Obstructive sleep apnea, chronic obstructive pulmonary disease and NAFLD: An individual participant data meta-analysis. Sleep Med. 2021, 77, 357–364.
- Savransky, V.; Bevans, S.; Nanayakkara, A.; Li, J.; Smith, P.L.; Torbenson, M.S.; Polotsky, V.Y. Chronic intermittent hypoxia causes hepatitis in a mouse model of diet-induced fatty liver. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G871–G877.
- Zhou, J.; Zhao, Y.; Guo, Y.J.; Zhao, Y.S.; Liu, H.; Ren, J.; Li, J.R.; Ji, E.S. A rapid juvenile murine model of nonalcoholic steatohepatitis (NASH): Chronic intermittent hypoxia exacerbates western diet-induced NASH. Life Sci. 2021, 276, 119403.
- Paschetta, E.; Belci, P.; Alisi, A.; Liccardo, D.; Cutrera, R.; Musso, G.; Nobili, V. OSAS-related inflammatory mechanisms of liver injury in nonalcoholic fatty liver disease. Mediat. Inflamm. 2015, 2015, 815721.
- Briançon-Marjollet, A.; Monneret, D.; Henri, M.; Joyeux-Faure, M.; Totoson, P.; Cachot, S.; Faure, P.; Godin-Ribuot, D. Intermittent hypoxia in obese Zucker rats: Cardiometabolic and inflammatory effects. Exp. Physiol. 2016, 101, 1432–1442.
- Uchiyama, T.; Ota, H.; Itaya-Hironaka, A.; Shobatake, R.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Makino, M.; Kimura, H.; Takeda, M.; Ohbayashi, C.; et al. Up-regulation of selenoprotein P and HIP/PAP mRNAs in hepatocytes by intermittent hypoxia via down-regulation of miR-203. Biochem. Biophys. Rep. 2017, 11, 130–137.
- Takasawa, S. Regenerating gene (REG) product and its potential clinical usage. Expert Opin. Ther. Targets 2016, 20, 541–550.
- Terazono, K.; Yamamoto, H.; Takasawa, S.; Shiga, K.; Yonemura, Y.; Tochino, Y.; Okamoto, H. A novel gene activated in regenerating islets. J. Biol. Chem. 1988, 263, 2111–2114.
- Okamoto, H.; Takasawa, S. Okamoto model for necrosis and its expansions, CD38-cyclic ADP-ribose signal system for intracellular Ca2+ mobilization and Reg ( protein)-Reg receptor system for cell regeneration. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2021, 79, 423–461.
- Shervani, N.J.; Takasawa, S.; Uchigata, Y.; Akiyama, T.; Nakagawa, K.; Noguchi, N.; Takada, H.; Takahashi, I.; Yamauchi, A.; Ikeda, T.; et al. Autoantibodies to REG, a beta-cell regeneration factor, in diabetic patients. Eur. J. Clin. Investig. 2004, 34, 752–758.
- Watanabe, T.; Yonemura, Y.; Yonekura, H.; Suzuki, Y.; Miyashita, H.; Sugiyama, K.; Moriizumi, S.; Unno, M.; Tanaka, O.; Kondo, H.; et al. Pancreatic beta-cell replication and amelioration of surgical diabetes by Reg protein. Proc. Natl. Acad. Sci. USA 1994, 91, 3589–3592.
- Ota, H.; Itaya-Hironaka, A.; Yamauchi, A.; Sakuramoto-Tsuchida, S.; Miyaoka, T.; Fujimura, T.; Tsujinaka, H.; Yoshimoto, K.; Nakagawara, K.; Tamaki, S.; et al. Pancreatic β cell proliferation by intermittent hypoxia via up-regulation of Reg family genes and HGF gene. Life Sci. 2013, 93, 664–672.
- Takeda, Y.; Itaya-Hironaka, A.; Yamauchi, A.; Makino, M.; Sakuramoto-Tsuchida, S.; Ota, H.; Kawaguchi, R.; Takasawa, S. Intermittent hypoxia upregulates the renin and Cd38 mRNAs in renin-producing cells via the downregulation of miR-203. Int. J. Mol. Sci. 2021, 22, 10127.
- Ryan, S. Adipose tissue inflammation by intermittent hypoxia: Mechanistic link between obstructive sleep apnoea and metabolic dysfunction. J. Physiol. 2017, 595, 2423–2430.
- Ryan, S.; Arnaud, C.; Fitzpatrick, S.F.; Gaucher, J.; Tamisier, R.; Pépin, J.L. Adipose tissue as a key player in obstructive sleep apnoea. Eur. Respir. Rev. 2019, 28, 190006.
- Jo, J.; Gavrilova, O.; Pack, S.; Jou, W.; Mullen, S.; Sumner, A.E.; Cushman, S.W.; Periwal, V. Hypertrophy and/or hyperplasia: Dynamics of adipose tissue growth. PLoS Comput. Biol. 2009, 5, e1000324.
- Jung, U.J.; Choi, M.S. Obesity and its metabolic complications: The role of adipokines and the relationship between obesity, inflammation, insulin resistance, dyslipidemia and nonalcoholic fatty liver disease. Int. J. Mol. Sci. 2014, 15, 6184–6223.
- Rajesh, Y.; Sarkar, D. Association of adipose tissue and adipokines with development of obesity-induced liver cancer. Int. J. Mol. Sci. 2021, 22, 2163.
- Trayhurn, P. Hypoxia and adipose tissue function and dysfunction in obesity. Physiol. Rev. 2013, 93, 1–21.
- Prabhakar, N.R.; Kumar, G.K. Mechanisms of sympathetic activation and blood pressure elevation by intermittent hypoxia. Respir. Physiol. Neurobiol. 2010, 174, 156–161.
- Li, Q.; Zhao, M.; Wang, Y.; Zhong, F.; Liu, J.; Gao, L.; Zhao, J. Associations between serum free fatty acid levels and incident diabetes in a 3-year cohort study. Diabetes Metab. Syndr. Obes. 2021, 14, 2743–2751.
- Lytrivi, M.; Castell, A.L.; Poitout, V.; Cnop, M. Recent insights into mechanisms of β-cell lipo- and glucolipotoxicity in Type 2 diabetes. J. Mol. Biol. 2020, 432, 1514–1534.
- Benito-Vicente, A.; Jebari-Benslaiman, S.; Galicia-Garcia, U.; Larrea-Sebal, A.; Uribe, K.B.; Martin, C. Molecular mechanisms of lipotoxicity-induced pancreatic β-cell dysfunction. Int. Rev. Cell Mol. Biol. 2021, 359, 357–402.
- Gileles-Hillel, A.; Almendros, I.; Khalyfa, A.; Nigdelioglu, R.; Qiao, Z.; Hamanaka, R.B.; Mutlu, G.M.; Akbarpour, M.; Gozal, D. Prolonged exposures to intermittent hypoxia promote visceral white adipose tissue inflammation in a murine model of severe sleep apnea: Effect of normoxic recovery. Sleep 2017, 40, zsw074.
- Gabryelska, A.; Karuga, F.F.; Szmyd, B.; Białasiewicz, P. HIF-1α as a mediator of insulin resistance, T2DM, and its complications: Potential links with obstructive sleep apnea. Front. Physiol. 2020, 11, 1035.
- Gabryelska, A.; Szmyd, B.; Panek, M.; Szemraj, J.; Kuna, P.; Białasiewicz, P. Serum hypoxia-inducible factor-1α protein level as a diagnostic marker of obstructive sleep apnea. Pol. Arch. Intern. Med. 2020, 130, 158–160.
- Ryan, S.; Taylor, C.T.; McNicholas, W.T. Selective activation of inflammatory pathways by intermittent hypoxia in obstructive sleep apnea syndrome. Circulation 2005, 112, 2660–2667.
- Korbecki, J.; Simińska, D.; Gąssowska-Dobrowolska, M.; Listos, J.; Gutowska, I.; Chlubek, D.; Baranowska-Bosiacka, I. Chronic and cycling hypoxia: Drivers of cancer chronic inflammation through HIF-1 and NF-κB activation: A review of the molecular mechanisms. Int. J. Mol. Sci. 2021, 22, 10701.
- Kawai, T.; Autieri, M.V.; Scalia, R. Adipose tissue inflammation and metabolic dysfunction in obesity. Am. J. Physiol. Cell Physiol. 2021, 320, C375–C391.
- Feijóo-Bandín, S.; Aragón-Herrera, A.; Moraña-Fernández, S.; Anido-Varela, L.; Tarazón, E.; Roselló-Lletí, E.; Portolés, M.; Moscoso, I.; Gualillo, O.; González-Juanatey, J.R.; et al. Adipokines and inflammation: Focus on cardiovascular diseases. Int. J. Mol. Sci. 2020, 21, 7711.
- Reddy, S.; Amutha, A.; Rajalakshmi, R.; Bhaskaran, R.; Monickaraj, F.; Rangasamy, S.; Anjana, R.M.; Abhijit, S.; Gokulakrishnan, K.; Das, A.; et al. Association of increased levels of MCP-1 and cathepsin-D in young onset type 2 diabetes patients (T2DM-Y) with severity of diabetic retinopathy. J. Diabetes Complicat. 2017, 31, 804–809.
- Chuang, L.P.; Chen, N.H.; Lin, Y.; Ko, W.S.; Pang, J.H. Increased MCP-1 gene expression in monocytes of severe OSA patients and under intermittent hypoxia. Sleep Breath. 2016, 20, 425–433.
- Barros, D.; García-Río, F. Obstructive sleep apnea and dyslipidemia: From animal models to clinical evidence. Sleep 2019, 42, 1–15.
- Drager, L.F.; Li, J.; Shin, M.K.; Reinke, C.; Aggarwal, N.R.; Jun, J.C.; Bevans-Fonti, S.; Sztalryd, C.; O’Byrne, S.M.; Kroupa, O.; et al. Intermittent hypoxia inhibits clearance of triglyceride-rich lipoproteins and inactivates adipose lipoprotein lipase in a mouse model of sleep apnoea. Eur. Heart J. 2012, 33, 783–790.
- de Lima, F.F.; Mazzotti, D.R.; Tufik, S.; Bittencourt, L. The role inflammatory response genes in obstructive sleep apnea syndrome: A review. Sleep Breath. 2016, 20, 331–338.
- Ge, M.Q.; Yeung, S.C.; Mak, J.C.W.; Ip, M.S.M. Differential metabolic and inflammatory responses to intermittent hypoxia in substrains of lean and obese C57BL/6 mice. Life Sci. 2019, 238, 116959.
- Akasaka, J.; Naruse, K.; Sado, T.; Uchiyama, T.; Makino, M.; Yamauchi, A.; Ota, H.; Sakuramoto-Tsuchida, S.; Itaya-Hironaka, A.; Takasawa, S.; et al. Involvement of receptor for advanced glycation endproducts in hypertensive disorders of pregnancy. Int. J. Mol. Sci. 2019, 20, 5462.
- Venkata, C.; Venkateshiah, S.B. Sleep-disordered breathing during pregnancy. J. Am. Board Fam. Med. 2009, 22, 158–168.
- Pengo, M.F.; Rossi, G.P.; Steier, J. Obstructive sleep apnea, gestational hypertension and preeclampsia: A review of the literature. Curr. Opin. Pulm. Med. 2014, 20, 588–594.
- Dominguez, J.E.; Street, L.; Louis, J. Management of obstructive sleep apnea in pregnancy. Obstet. Gynecol. Clin. N. Am. 2018, 45, 233–247.
- Ota, H.; Fujita, Y.; Yamauchi, M.; Muro, S.; Kimura, H.; Takasawa, S. Relationship between intermittent hypoxia and Type 2 diabetes in sleep apnea syndrome. Int. J. Mol. Sci. 2019, 20, 4756.
- Oh, K.J.; Lee, D.S.; Kim, W.K.; Han, B.S.; Lee, S.C.; Bae, K.H. Metabolic adaptation in obesity and Type II diabetes: Myokines, adipokines and hepatokines. Int. J. Mol. Sci. 2016, 18, 8.
- Laurens, C.; Bergouignan, A.; Moro, C. Exercise-released myokines in the control of energy metabolism. Front. Physiol. 2020, 11, 91.
- Di Felice, V.; Coletti, D.; Seelaender, M. Editorial: Myokines, adipokines, cytokines in muscle pathophysiology. Front. Physiol. 2020, 11, 592856.
- Wedell-Neergaard, A.S.; Lang Lehrskov, L.; Christensen, R.H.; Legaard, G.E.; Dorph, E.; Larsen, M.K.; Launbo, N.; Fagerlind, S.R.; Seide, S.K.; Nymand, S.; et al. Exercise-induced changes in visceral adipose tissue mass are regulated by IL-6 signaling: A randomized controlled trial. Cell Metab. 2019, 29, 844–855.
- Park, T.J.; Park, A.; Kim, J.; Kim, J.Y.; Han, B.S.; Oh, K.J.; Lee, E.W.; Lee, S.C.; Bae, K.H.; Kim, W.K. Myonectin inhibits adipogenesis in 3T3-L1 preadipocytes by regulating p38 MAPK pathway. BMB Rep. 2021, 54, 124–129.
- Little, H.C.; Rodriguez, S.; Lei, X.; Tan, S.Y.; Stewart, A.N.; Sahagun, A.; Sarver, D.C.; Wong, G.W. Myonectin deletion promotes adipose fat storage and reduces liver steatosis. FASEB J. 2019, 33, 8666–8687.
- Eckel, J. Myokines in metabolic homeostasis and diabetes. Diabetologia 2019, 62, 1523–1528.
- Garneau, L.; Aguer, C. Role of myokines in the development of skeletal muscle insulin resistance and related metabolic defects in type 2 diabetes. Diabetes Metab. 2019, 45, 505–516.
- Atakan, M.M.; Koşar, Ş.N.; Güzel, Y.; Tin, H.T.; Yan, X. The role of exercise, diet, and cytokines in preventing obesity and improving adipose tissue. Nutrients 2021, 13, 1459.
- Otaka, N.; Shibata, R.; Ohashi, K.; Uemura, Y.; Kambara, T.; Enomoto, T.; Ogawa, H.; Ito, M.; Kawanishi, H.; Maruyama, S.; et al. Myonectin is an exercise-induced myokine that protects the heart from ischemia-reperfusion injury. Circ. Res. 2018, 123, 1326–1338.
- Yin, X.; Zheng, Y.; Zhai, X.; Zhao, X.; Cai, L. Diabetic inhibition of preconditioning- and postconditioning-mediated myocardial protection against ischemia/reperfusion injury. Exp. Diabetes Res. 2012, 2012, 198048.
- Szabó, M.R.; Pipicz, M.; Csont, T.; Csonka, C. Modulatory effect of myokines on reactive oxygen species in ischemia/reperfusion. Int. J. Mol. Sci. 2020, 21, 9382.
- Takasawa, S.; Shobatake, R.; Itaya-Hironaka, A.; Makino, M.; Sakuramoto-Tsuchida, S.; Uchiyama, T.; Ota, H.; Yamauchi, A. Up-regulation of IL-8, osteonectin, and myonectin mRNAs by intermittent hypoxia via OCT1- and NRF2-mediated mechanisms in skeletal muscle cells. Diabetologia 2020, 63 (Suppl. S1), 453.
- Maciel, L.; de Oliveira, D.F.; Mesquita, F.; Souza, H.; Oliveira, L.; Christie, M.L.A.; Palhano, F.L.; Campos de Carvalho, A.C.; Nascimento, J.H.M.; Foguel, D. New cardiomyokine reduces myocardial ischemia/reperfusion injury by PI3K-AKT pathway via a putative KDEL-receptor binding. J. Am. Heart Assoc. 2021, 10, e019685.
- Maciel, L.; de Oliveira, D.F.; Verissimo da Costa, G.C.; Bisch, P.M.; Nascimento, J.H.M. Cardioprotection by the transfer of coronary effluent from ischaemic preconditioned rat hearts: Identification of cardioprotective humoral factors. Basic Res. Cardiol. 2017, 112, 52.
- Glembotski, C.C. Functions for the cardiomyokine, MANF, in cardioprotection, hypertrophy and heart failure. J. Mol. Cell. Cardiol. 2011, 51, 512–517.
- Takasawa, S.; Itaya-Hironaka, A.; Yamauchi, A.; Makino, M.; Sakuramoto-Tsuchida, S.; Uchiyama, T.; Takeda, Y.; Kyotani, Y.; Ota, H. Up-regulation of regenerating gene IV and hepatocyte growth factor in cardiomyocytes by intermittent hypoxia and its microRNA-mediated mechanism. Diabetes 2021, 70 (Suppl. S1), 378-P.
This entry is offline, you can click here to edit this entry!